


Os autores apresentam um raro caso de miocardiopatia hipertrófica (MCH) associada a ventrículo esquerdo não compactado (VENC) e fístulas das artérias coronárias para o ventrículo esquerdo (VE) numa doente de 42 anos, que se apresenta com enfarte agudo do miocárdio sem elevação do segmento ST (EAMSEST). A coronariografia, a ecocardiografia transtorácica (ETT) e ressonância magnética cardíaca (RMC) revelaram as alterações estruturais do VE e da árvore coronária.
The authors present a rare case of hypertrophic cardiomyopathy associated with left ventricular noncompaction cardiomyopathy and coronary artery-left ventricular fistulae in a 42-year-old woman presenting with non-ST-elevation myocardial infarction. Coronary angiography, transthoracic echocardiography and cardiac magnetic resonance revealed the structural abnormalities of the left ventricle and the coronary tree.
A miocardiopatia hipertrófica (MCH) é a doença cardíaca genética mais comum, com uma prevalência de 1:5001. Embora associada a uma heterogeneidade fenotípica2,3, a MCH caracteriza-se pela presença de hipertrofia do ventrículo esquerdo (VE) com função sistólica preservada e relaxamento diminuído, na ausência de outra causa identificável4. É herdada de forma autossómica dominante e até ao momento mais de 900 mutações diferentes foram relatadas em cerca de 11 genes que codificam proteínas sarcoméricas5. As mutações mais frequentes são as que envolvem a β-miosina de cadeia pesada (MYH7), a troponina T (TNNT2) e a proteína-C ligada à miosina6.
Na histopatologia, observa-se um desarranjo das fibras miocárdicas e fibrose4.
O diagnóstico clínico é difícil, particularmente nas formas não obstrutivas. Os critérios ecocardiográficos, clássicos, utilizados no diagnóstico de MCH são: espessura máxima da parede superior a 15mm em qualquer segmento do miocárdio (predominando o atingimento assimétrico do septo); função sistólica global normal com disfunção diastólica; presença de gradiente intraventricular; movimento anterior sistólico mitral e insuficiência mitral; aspeto granuloso do miocárdio2,4,7,8. A avaliação da extensão e gravidade da hipertrofia deve incluir a medição da espessura máxima da parede em vários segmentos7. O diagnóstico clínico de MCH também pode ser reforçado por outras características típicas, como a história familiar de doença cardíaca, dispneia de esforço, angor, síncope, taquiarritmias, insuficiência mitral ou alterações electrocardiográficas9. O teste genético possibilita o rastreio de familiares portadores de mutação, mas não prevê o curso clínico de cada paciente, embora este possa ser influenciado por mutações específicas6,10.
O ventrículo esquerdo não compactado (VENC) é uma cardiomiopatia rara, congénita, mas por vezes adquirida11, com uma prevalência inferior a 1:5.00012. A interrupção do processo de compactação miocárdica durante a embriogénese é uma das teorias explicativas da VENC.
Pelo final da quarta semana de gestação fetal surgem invaginações endocárdicas que cruzam a «geleia» embrionária cardíaca até ao miocárdio adjacente. Neste, por sua vez, desenvolvem-se formações sinusoidais ou recessos, relacionados posteriormente com a circulação coronária13. O rendilhado e protusão de trabeculações no lúmen ventricular conferem ao coração o aspeto de uma esponja. Entre a 12.a-18.a semanas de gestação ocorre uma compactação gradual do miocárdio que progride da base do coração para o ápex12,14. Este processo é mediado através de vários fatores de crescimento neuro-humorais e angiogénicos15.
Neste tipo de miocardiopatia (VENC) poderão ser considerados dois subgrupos: um caracterizado pela ausência de outras anomalias cardíacas congénitas concomitantes e outro onde a coexistência das referidas anomalias, incluindo microfístulas coronárias, estão presentes16. Os aspetos morfológicos identificados na ecocardiografia são trabeculações excessivas do VE, com uma relação entre as camadas não compactada-compactada na tele-sístole ≥2:1 e recessos endomiocárdicos que comunicam com a cavidade ventricular17. As características do VENC atingem predominantemente a região apical e segmentos médio-distais póstero-laterais do VE18. Tipicamente estes segmentos são hipocinéticos, causando disfunção sistólica17. Recentemente, mutações nos genes que codificam proteínas sarcoméricas, as quais têm sido anteriormente implicadas na patogénese da MCH, foram identificadas em pacientes com VENC19,20. A sintomatologia é inespecífica desde insuficiência cardíaca, alterações da condução e eventos tromboembólicos associados à formação de trombos nos recessos intertrabeculares15. O prognóstico é variável dada a natureza heterogénea desta miocardiopatia21.
Apresentação de casoRelatamos o caso de uma mulher de 42 anos de idade, admitida no serviço de urgência com desconforto precordial, palpitações e dispneia com duas horas de evolução. Apresentava antecedentes pessoais de MCH, fibrilhação auricular permanente e acidente vascular cerebral em 2002. Nessa altura fez ecocardiograma transtorácico que revelava SIV hipertrofiado com cerca de 18mm de espessura, sem gradiente intraventricular, aurícula esquerda dilatada (42mm), ventrículo esquerdo com função sistólica global preservada, não existindo imagens sugestivas de trombos intracardíacos. Encontrava-se agora medicada com carvedilol 6,25mg bid, digoxina 0,25mg id e varfarina para INR 2-3. Tinha história familiar de MCH – um irmão de 40 anos de idade afetado e um irmão e uma irmã falecidos de morte súbita cardíaca aos 24 e 46 anos, respetivamente. O exame objetivo não revelou alterações, além de auscultação cardíaca arrítmica. O eletrocardiograma mostrou fibrilhação auricular com resposta ventricular controlada e perturbações inespecíficas da condução intraventricular. A radiografia de tórax evidenciou cardiomegalia sem edema pulmonar. Analiticamente apresentava elevação da troponina I (5,2ng/ml) e hipercolesterolémia. Foi internada com diagnóstico de EAMSEST.
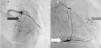
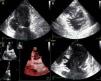
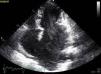
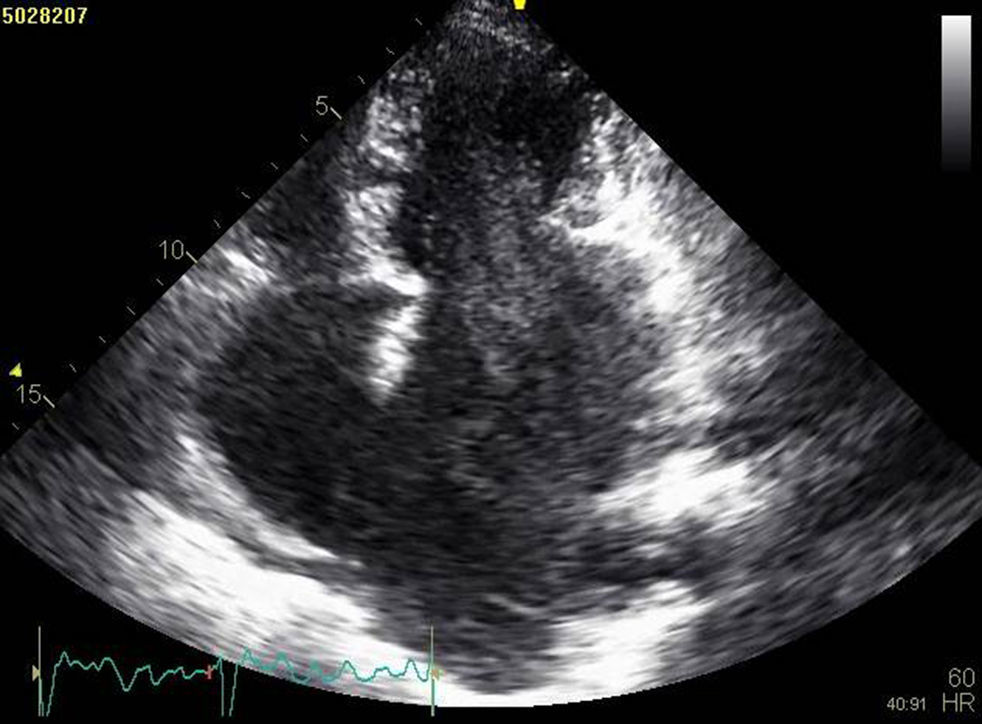
A doente foi submetida a cateterismo cardíaco, a fim de excluir doença coronária. A coronariografia revelou a presença de bridging em várias localizações da artéria coronária esquerda (descendente anterior média, ramos septais e ramos distais da primeira diagonal), também se identificando várias fístulas das coronárias esquerda e direita para o ventrículo esquerdo. Visualizava-se um trombo recanalizado no ramo póstero-lateral (fig. 1). O ecocardiograma bidimensional transtorácico mostrou hipertrofia ventricular esquerda assimétrica envolvendo principalmente o septo interventricular (22mm) e a parede lateral. Observava-se um aneurisma basal póstero-inferior. A função sistólica global encontrava-se preservada (∼55% por Simpson biplano). Na porção basal da parede septal inferior eram visíveis dois recessos intramiocárdicos em comunicação com a cavidade do VE, com cerca de 4mm de diâmetro, o maior dos quais atravessando quase toda a espessura da parede, na ausência de comunicação interventricular (fig. 2). A aurícula esquerda encontrava-se severamente dilatada, com contraste espontâneo nas cavidades esquerdas (fig. 3).
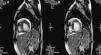
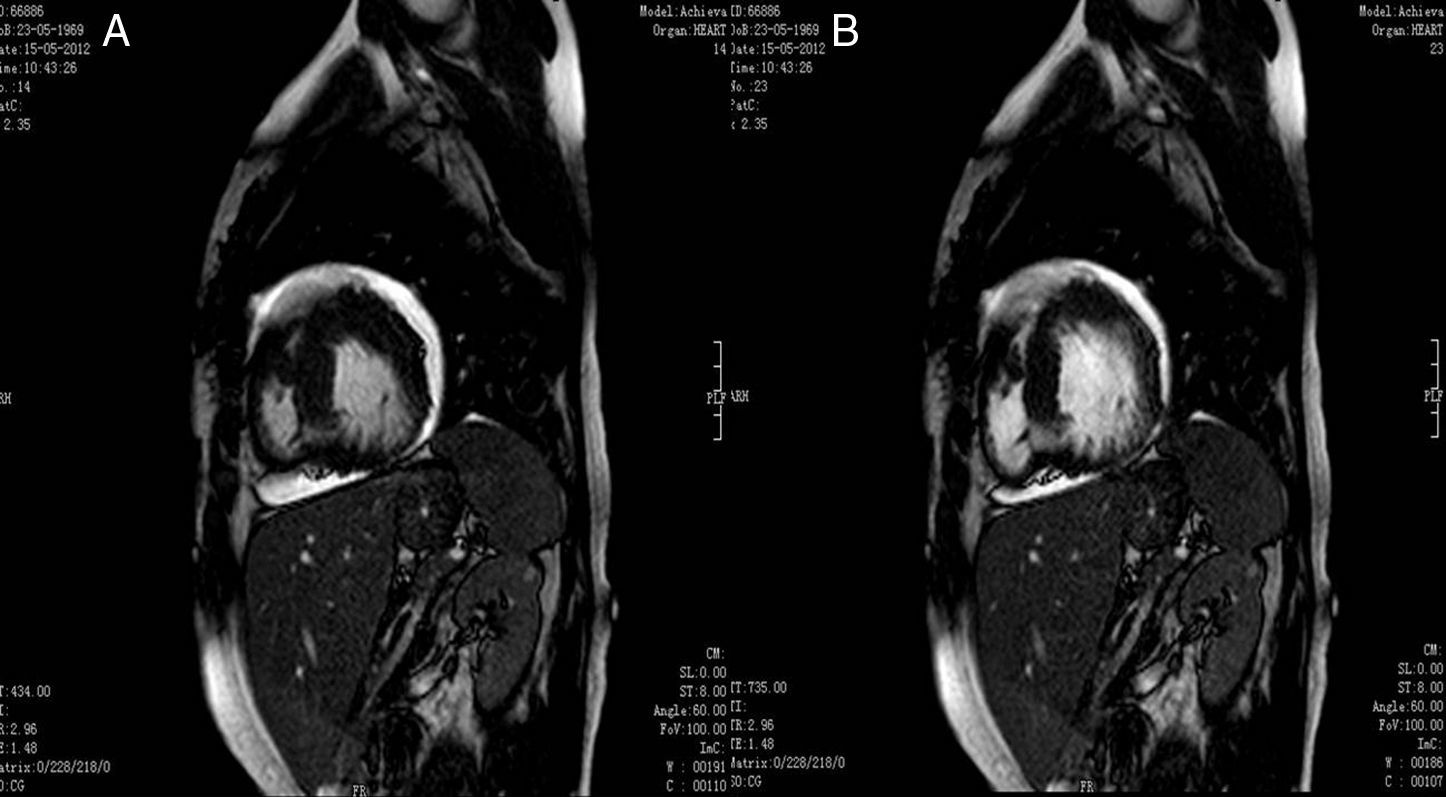
A fim de corroborar o diagnóstico foi realizada uma RMC que revelou uma relação de 2,7 entre as camadas não-compactada e compactada, bem como imagens sugestivas de recessos intramurais e função sistólica do ventrículo esquerdo preservada, conforme anteriormente descrito na ecocardiografia transtorácica (ETT) (fig. 4).
O estudo genético identificou a mutação c.1208G> A no exão 13 do gene MYH7.
Após um internamento de 12 dias, a doente teve alta medicada com metoprolol 50mg b.i.d, atorvastatina 10mg id, varfarina e furosemida 40mg id.
Decorrido um mês, procedeu-se à implantação de um CDI. Posteriormente, uma descompensação da insuficiência cardíaca motivou um reinternamento, de curta duração, estando estável desde então.
DiscussãoApresentamos um caso, raro, que combina o fenótipo de duas miocardiopatias (MCH e VENC) com múltiplas fístulas coronário-ventriculares e recessos intramiocárdicos que, provavelmente, correspondem a grandes vasos sinusoidais.
Supõe-se que a coexistência de fístulas coronárias, de recessos intramiocárdicos e de miocárdio esponjoso resulte de uma anomalia no desenvolvimento embrionário, alterando a regressão dos sinusoides e a compactação do miocárdio traduzindo uma doença só22–24; a camada compactada dá origem ao miocárdio ventricular e a interrupção da compactação resulta na persistência de recessos intertrabeculares e na formação, incompleta, de capilares13,17.
Contrariamente às fístulas arteriovenosas ou arterio-arteriais, as fístulas coronário-ventriculares são extremamente raras, sendo mais frequentes no género feminino e na maioria congénitas25. A importância clínica depende da dimensão e localização da fístula, sendo na maioria um achado incidental e assintomático26.
Raramente foram identificadas múltiplas fístulas coronárias-VE em casos de MCH apical27–29. As fístulas podem estar associadas à presença de hipertrofia regional, embora não seja claro se esta é causa ou efeito de anomalias da anatomia coronária27. Os mecanismos de isquemia miocárdica na MCH incluem um aumento das necessidades de oxigénio pelo miocárdio ventricular hipertrofiado, uma diminuição da circulação coronária e elevadas pressões de enchimento diastólico. As fístulas coronárias e o consequente fenómeno de roubo coronário e shunt esquerdo-esquerdo agravam a perfusão ventricular e aumentam a sobrecarga de volume diastólico23,29. No caso apresentado, apesar do grande número de fístulas, a função sistólica do VE está preservada.
Determinados estudos revelam que a presença de uma mesma mutação de um gene que codifica uma proteína sarcomérica (exemplo: MYH7 Arg243His) pode desencadear diferentes padrões patológicos de remodelação do miocárdio, nomeadamente MCH e VENC. Estas variações na expressão fenotípica poderão refletir uma interação entre o genótipo e os fatores ambientais20,30. A mutação identificada nesta família (Arg403Gln) está associada a MCH, mas não a VENC20. Foi estimado que cerca de 50% dos indivíduos portadores desta mutação apresentam MSC antes dos 50 anos de idade31. Um aspeto interessante deste caso prende-se com o facto de um irmão vivo da doente apresentar a mesma mutação, mas não ter desenvolvido VENC nem fístulas coronário-ventriculares, o que favorece a hipótese de etiologias diferentes para as duas miocardiopatias coexistentes. Assim, admitimos a origem genética da MCH e o mecanismo de interrupção embriológica do VENC.
Relativamente ao aneurisma póstero-inferior observado na ETT, não conseguimos definir a sua etiologia nem os exames prévios nos permitem esclarecer o momento do seu desenvolvimento.
O tromboembolismo arterial sistémico é uma complicação das miocardiopatias, especialmente quando associadas a aurícula esquerda dilatada e fibrilhação auricular32.
A fonte de embolia sistémica associada a MCH relaciona-se com a presença de trombos intracardíacos causados pela estase de fluxo sanguíneo, a nível da aurícula dilatada ou de aneurismas ventriculares33.
A incidência de eventos tromboembólicos varia de 5 a 38% em pacientes com VENC34.
Os mecanismos que contribuem para o desenvolvimento de trombose coronária em pessoas com fístulas coronárias incluem alterações do fluxo sanguíneo associadas à ectasia vascular, tortuosidade coronária, mudança abrupta no calibre dos vasos, alterações intrínsecas na cascata de coagulação ou fibrinólise e efeitos do trauma vascular35.
Relativamente ao caso em questão é de admitir a ocorrência de três possíveis mecanismos como causa do EAMSST: (1) evento embólico decorrente da fibrilhação auricular; (2) trombose intracoronária no contexto de fístulas coronárias – esta provavelmente devido aos fenómenos previamente descritos; e (3) bridging coronário, este último por redução de perfusão coronária quando existe atraso de relaxamento da parede arterial no início da diástole. O facto de a doente ter antecedentes pessoais de AVC isquémico mostra a existência de um risco elevado de novos eventos tromboembólicos.
Para nosso conhecimento, o presente caso é o primeiro diagnosticado in vivo que combina duas miocardiopatias e múltiplas fístulas coronário-ventriculares e enfatiza o papel complementar de diferentes técnicas de imagem na avaliação completa de tais anomalias cardiovasculares (Figuras 1–4).
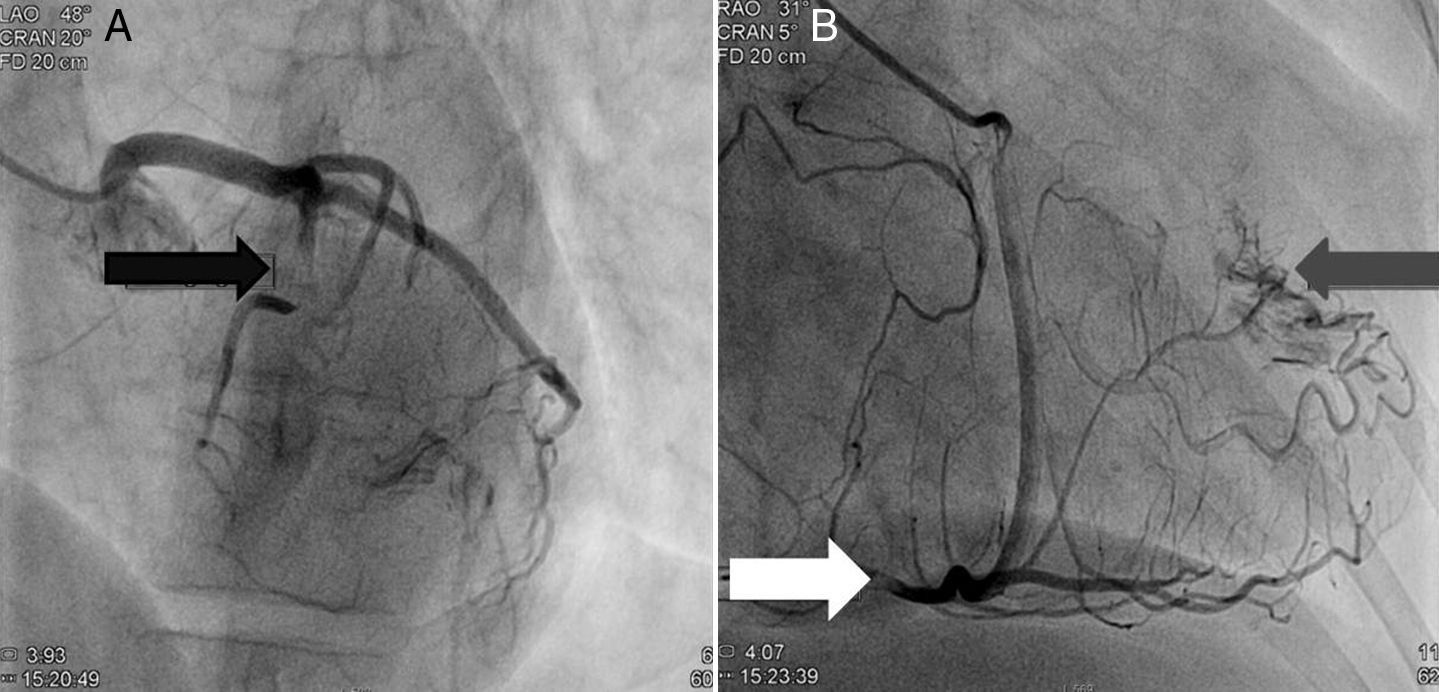
Coronariografia (A): bridging em várias localizações da artéria coronária esquerda (descendente anterior - seta preta). (B): identificam-se várias fístulas da coronária esquerda e coronária direita para o ventrículo esquerdo (seta cinzenta). Visualizava-se um trombo recanalizado no ramo póstero-lateral (seta branca).
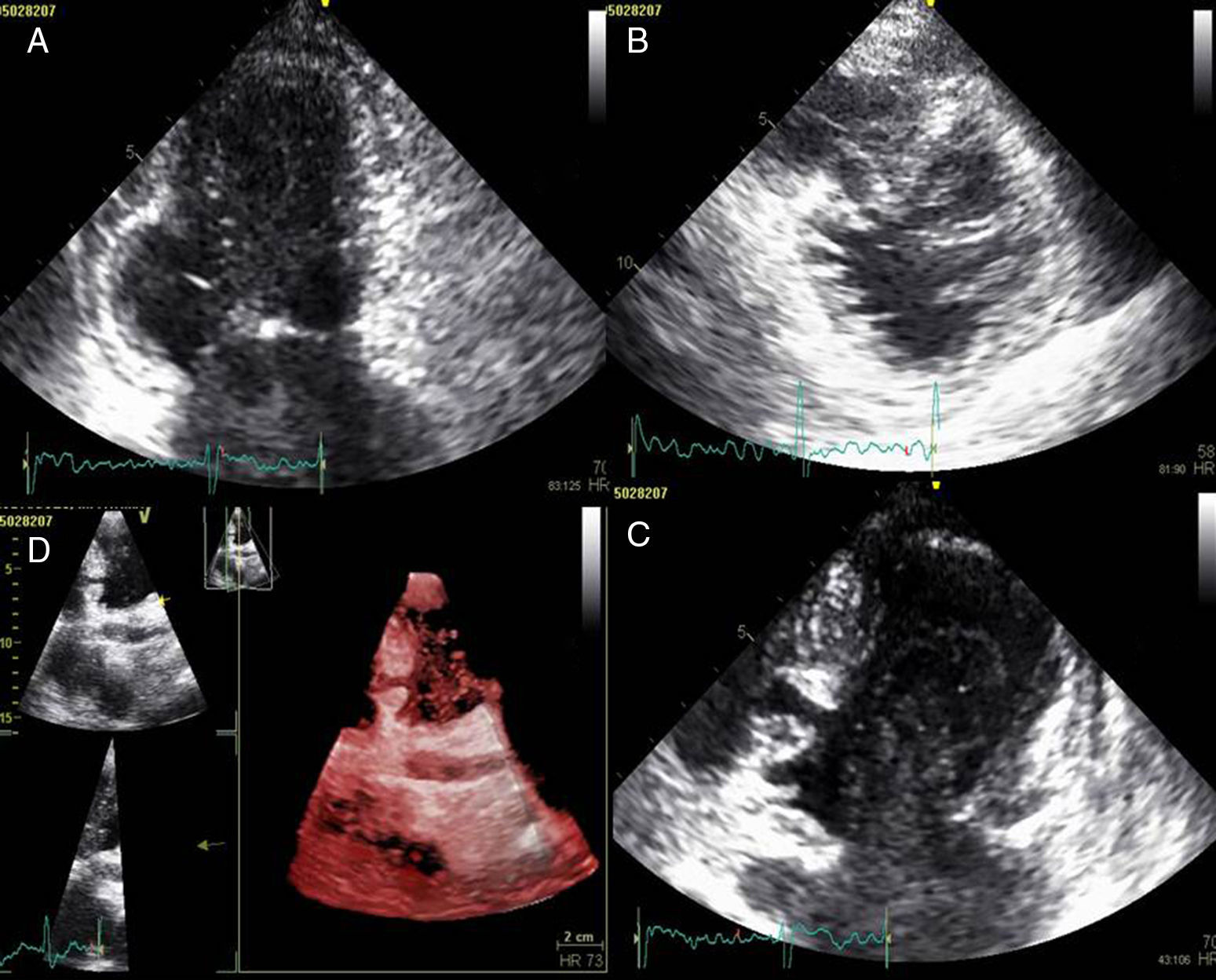
ETT (A): observa-se um aneurisma basal póstero-inferior (2D, plano apical duas câmaras). (B): são visíveis trabeculações e recessos endomiocárdicos em comunicação com a cavidade ventricular (2D, plano paraesternal, curto eixo). (C e D): na porção basal da parede septal inferior eram visíveis dois recessos intramiocárdicos em comunicação com a cavidade do VE, com cerca de 4mm de diâmetro, o maior dos quais atravessando quase toda a espessura da parede, na ausência de comunicação interventricular (imagem 2D e 3D, respetivamente, plano apical quatro câmaras).
Os autores declaram que para esta investigação não se realizaram experiências em seres humanos e/ou animais.
Confidencialidade dos dadosOs autores declaram ter seguido os protocolos de seu centro de trabalho acerca da publicação dos dados de pacientes e que todos os pacientes incluídos no estudo receberam informações suficientes e deram o seu consentimento informado por escrito para participar nesse estudo.
Direito à privacidade e consentimento escritoOs autores declaram ter recebido consentimento escrito dos pacientes e/ou sujeitos mencionados no artigo. O autor para correspondência deve estar na posse deste documento.
Conflito de interessesOs autores declaram não haver conflito de interesses.
