





Um homem de 67 anos deu entrada no hospital após episódios de síncope precedidos por mal‐estar geral e desconforto torácico difuso. Sem história familiar de doença cardíaca. O estudo analítico, incluindo d‐dímeros e troponina I, bem como a gasimetria arterial, não mostrou alterações. O eletrocardiograma exibiu um ritmo sinusal com inversão da onda T de V1‐V3. A angioTC foi negativa para tromboembolismo pulmonar.
O ecocardiograma transtorácico (ETT) mostrou uma dilatação marcada do ventrículo direito com septo interauricular intacto, sem hipertensão arterial pulmonar.
A ressonância magnética cardíaca (RMC) confirmou a dilatação ventricular direita e revelou hipocinesia marcada/acinesia da sua parede lateral. A prova de esforço foi negativa para isquemia.
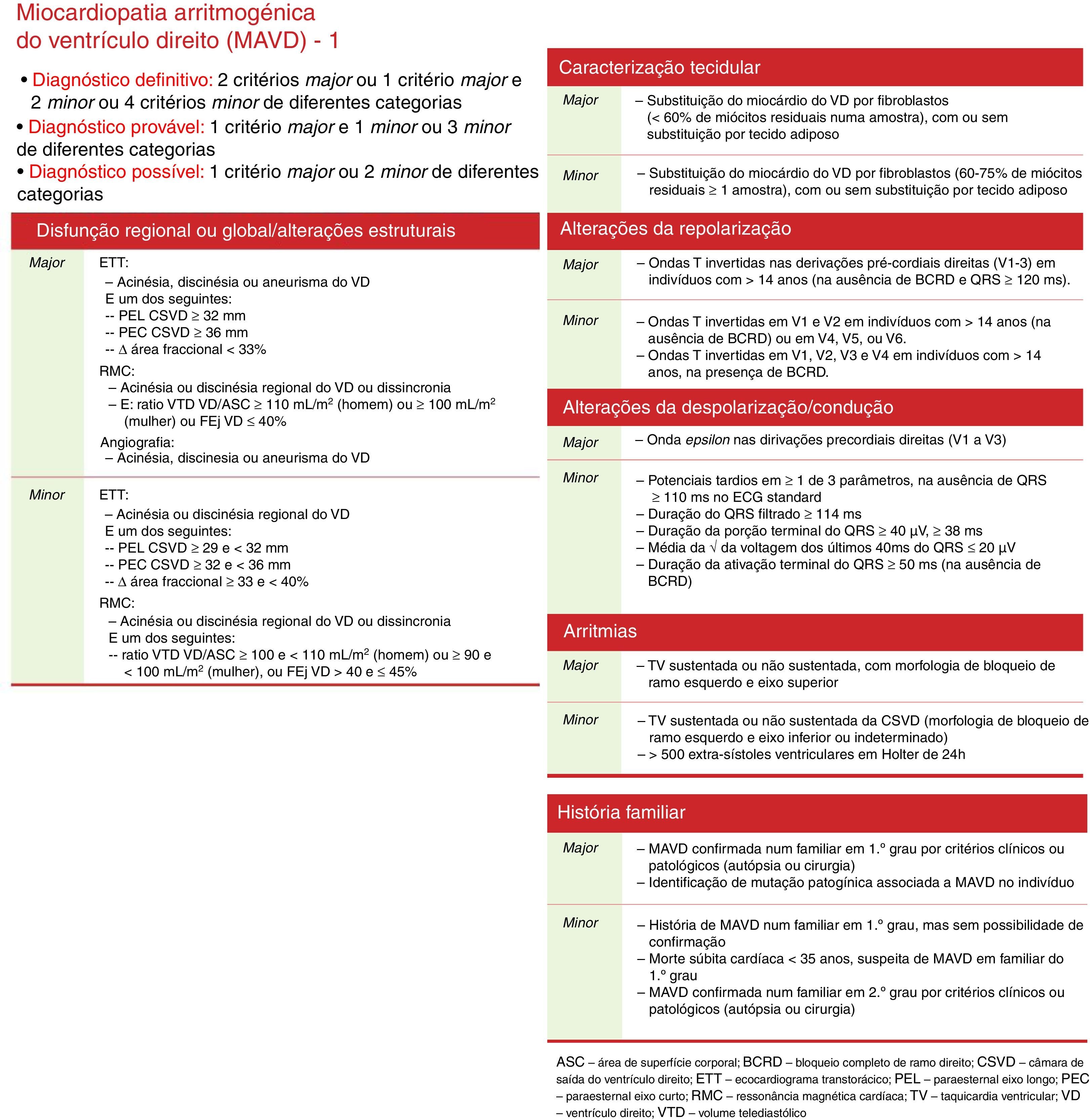
À luz dos critérios de diagnóstico de displasia arritmogénica do ventrículo direito (DAVD), preconizados pela Task Force (TF) de 2010, este doente apresentava dois critérios major (na RMC: acinesia ou discinesia regional do VD e fração de ejeção≤40%; no eletrocardiograma: ondas T invertidas nas derivações pré‐cordiais direitas [V1‐3]) e um minor (no electrocardiograma dinâmico de 24 horas [ECGD/Holter 24 horas]>500 extrassístoles ventriculares), pelo que o diagnóstico definitivo foi assumido.
Após o estudo eletrofisiológico, o doente foi submetido a colocação de cardioversor‐desfibrilhador implantável (CDI) monocâmara, encontrando‐se assintomático desde então.
Esta apresentação tardia de DAVD evidencia a importância do rastreio por ETT, complementado por RMC. O risco de morte súbita cardíaca foi aferido pelo estudo eletrofisiológico, conduzindo à implantação do CDI. Estudos de associação genética devem ser oferecidos aos descendentes do paciente.
A 67‐year‐old man was admitted to our hospital after episodes of syncope preceded by malaise and diffuse neck and chest discomfort. No family history of cardiac disease was reported. Laboratory workup was within normal limits, including D‐dimers, serum troponin I and arterial blood gases. The electrocardiogram showed sinus rhythm with T‐wave inversion in leads V1 to V3. Computed tomography angiography to investigate pulmonary embolism showed no abnormal findings. Transthoracic echocardiography (TTE) displayed massive enlargement of the right ventricle with intact interatrial septum and no pulmonary hypertension. Cardiac magnetic resonance imaging (MRI) confirmed right ventricular (RV) dilatation and revealed marked hypokinesia/akinesia of the lateral wall. Exercise stress testing was negative for ischemia.
According to the 2010 Task Force criteria for arrhythmogenic right ventricular dysplasia (ARVD), this patient presented two major criteria (global or regional dysfunction and structural alterations: by MRI, regional RV akinesia or dyskinesia or dyssynchronous RV contraction and RV ejection fraction ≤40%, and repolarization abnormalities: inverted T waves in right precordial leads [V1, V2, and V3]); and one minor criterion (>500 ventricular extrasystoles per 24 hours by Holter), and so a diagnosis of ARVD was made.
After electrophysiologic study (EPS) the patient received an implantable cardioverter‐defibrillator (ICD).
This late clinical presentation of ARVD highlights the importance of TTE screening, possibly complemented by MRI. The associated risk of sudden death was assessed by EPS leading to the implantation of an ICD. Genetic association studies should be offered to the offspring of all ARVD patients.
Um homem de 67 anos, autónomo nas atividades da vida diária, ex‐piloto de aviões, com antecedentes pessoais de hipertensão arterial essencial e dislipidemia (medicadas), tabagismo ativo (75UMA), sem antecedentes familiares relevantes, recorreu ao serviço de urgência de um hospital por episódio de mal‐estar geral, acompanhado de sensação de constrição cervical anterior e desconforto torácico discreto, seguidos de perda de conhecimento súbita com recuperação espontânea, completa e imediata. Negava dor precordial típica em aperto, dispneia, diaforese, náuseas e vómitos, astenia, alteração do volume dos membros inferiores, sintomas neurológicos focais, evidência de atividade convulsiva ou perda de continência de esfíncteres. Sem nova sintomatologia após este quadro, mantendo apenas sensação de mal‐estar geral.
Documentaram‐se troponina I e d‐dímeros negativos. A gasimetria arterial, radiografia postero‐anterior de tórax, eletrocardiograma (ECG), angioTC e TC cranioencefálica não revelaram alterações patológicas.
De acordo com o doente, admitiu‐se hipótese diagnóstica de síndrome coronária aguda inicialmente, ficando em observação durante 24 horas e tendo tido alta médica na manhã seguinte, sem indicação do diagnóstico definitivo.
Vem ao nosso serviço de urgência 24 horas depois, por quadro idêntico ao anteriormente descrito, exceto perda de conhecimento. Referiu, adicionalmente, presença de palpitações com duração de alguns segundos.
Negava outros sintomas e referiu história de outros episódios semelhantes, algumas semanas antes.
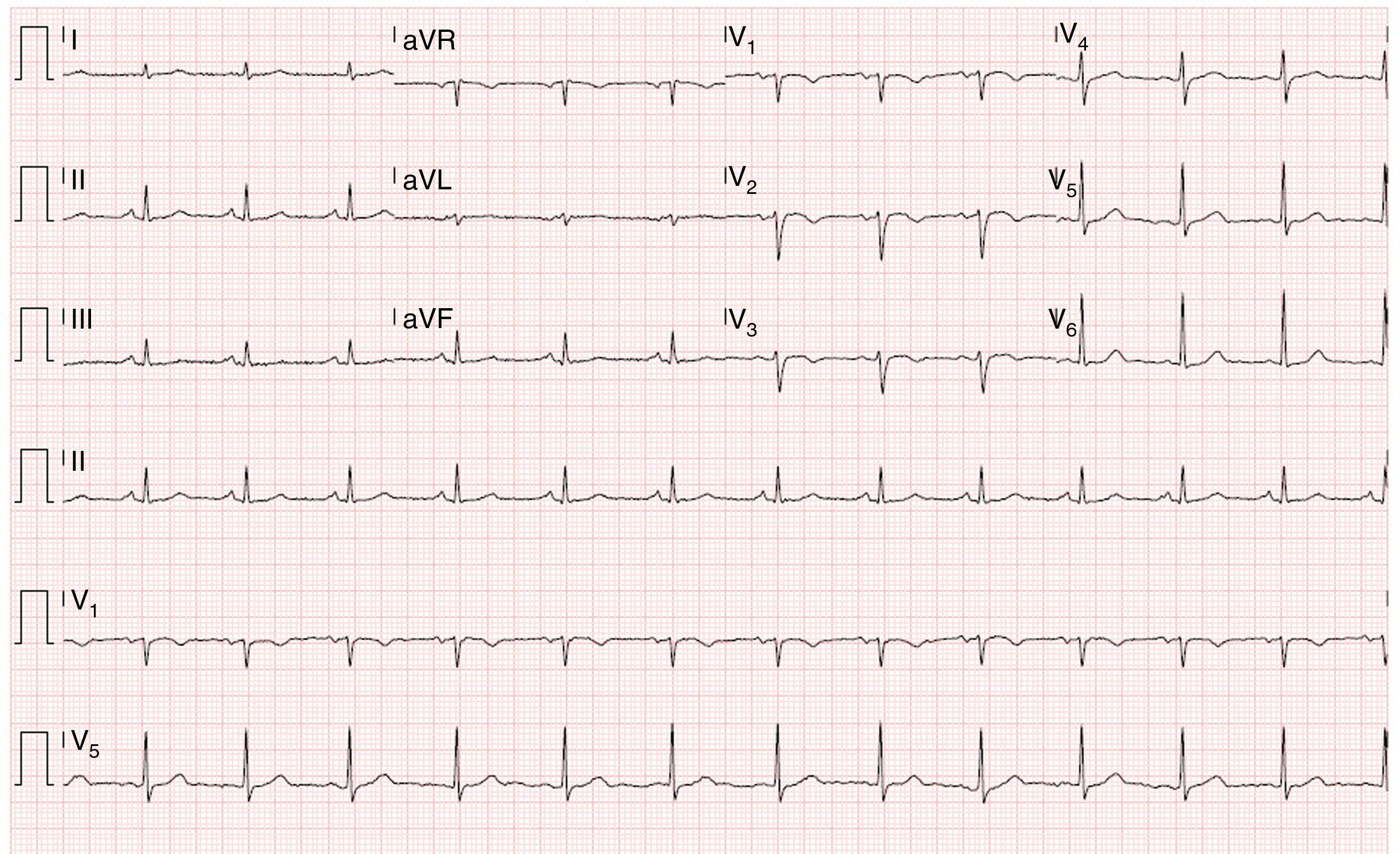
O exame objetivo foi normal. O ECG mostrou um traçado em ritmo sinusal, sem sinais de isquemia aguda, com inversão da onda T de V1‐V3 e ausência de onda épsilon (Figura 1).
A radiografia postero‐anterior de tórax não apresentou alterações relevantes.
Analiticamente, apresentava um hemograma sem alterações, sem elevação dos parâmetros de fase aguda, com função renal, ionograma, provas de coagulação, troponina I e CK‐MB com valores dentro dos limites da normalidade.
Foi realizado um ecocardiograma transtorácico que demonstrou um ventrículo esquerdo (VE) não dilatado, com boa função sistólica global, sem alterações da contractilidade segmentar e com fração de ejeção (FE) de 64%, e um ventrículo direito (VD) muito dilatado com tricuspid annular plane systolic excursion (TAPSE) de 19mm, aurícula direita dilatada, veia cava inferior com colapso inspiratório<50% e pressão sistólica da artéria pulmonar (PSAP) de 31,60mmHg. Sem outras alterações relevantes.
Durante o internamento o paciente evoluiu assintomático, analiticamente documentou‐se um valor de NT‐proBNP de 705pg/mL, com restante estudo sem alterações.
Perante a perda de conhecimento de etiologia a esclarecer, na presença de VD dilatado sem aparentes alterações da contractilidade e função ventriculares, foram pedidos ECGD/Holter 24 horas, prova de esforço e ressonância magnética cardíaca (RMC).
No ECGD/Holter 24 horas destacavam‐se uma média de 22 sístoles prematuras ventriculares por hora (528 nas 24 horas), bimorfas, isoladas e seis pares. A prova de esforço, com protocolo de Bruce, teve uma duração de sete minutos e 30 segundos e foi terminada por fadiga do doente, tendo‐se atingido 90% da frequência máxima prevista para a idade. Durante todo o seu curso o doente negou angina e não se registaram alterações hemodinâmicas ou eletrocardiográficas patológicas.
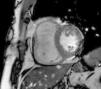
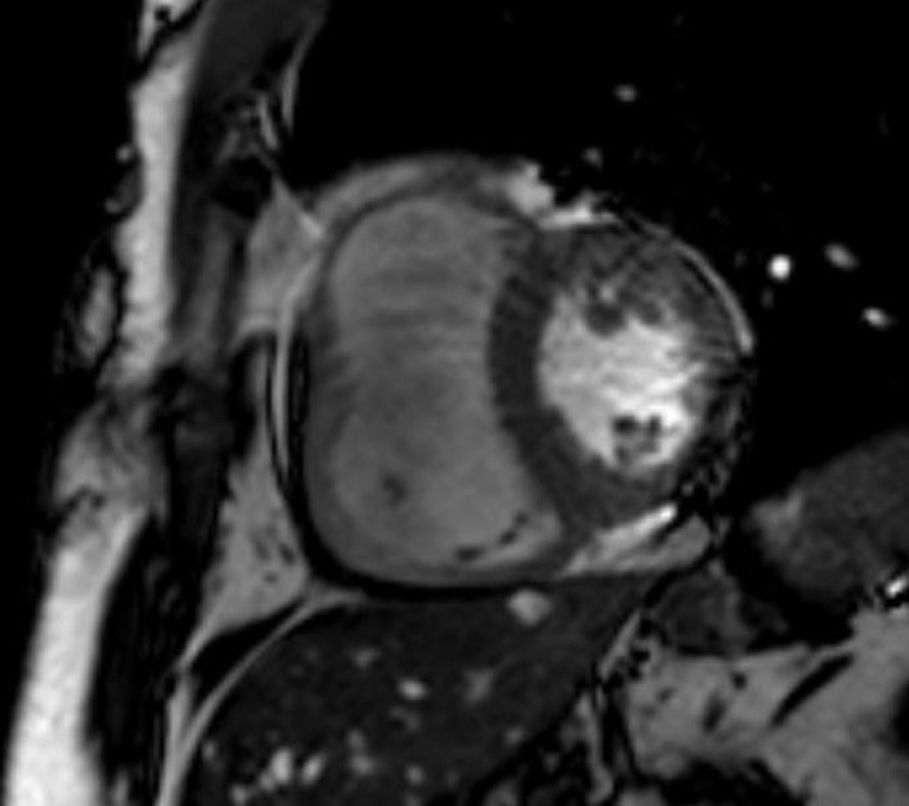
A RMC apresentou um VD dilatado com hipocinesia muito marcada/acinesia da parede livre e uma FE de 21%. Sem realce tardio após injeção de contraste paramagnético ao longo do miocárdio (Figura 2).
Baseado nestes achados, foi assumido estarem cumpridos dois critérios major e um minor para o diagnóstico definitivo de miocardiopatia/displasia arritmogénica do ventrículo direito (DAVD), segundo a Fask Force 2010 (Figura 3), nomeadamente, na RMC – acinesia ou discinesia regional do VD e FE≤40%, no ECG – ondas T invertidas nas derivações pré‐cordiais direitas (V1‐3) e no ECGD/Holter>500 extrassístoles ventriculares nas 24 horas.
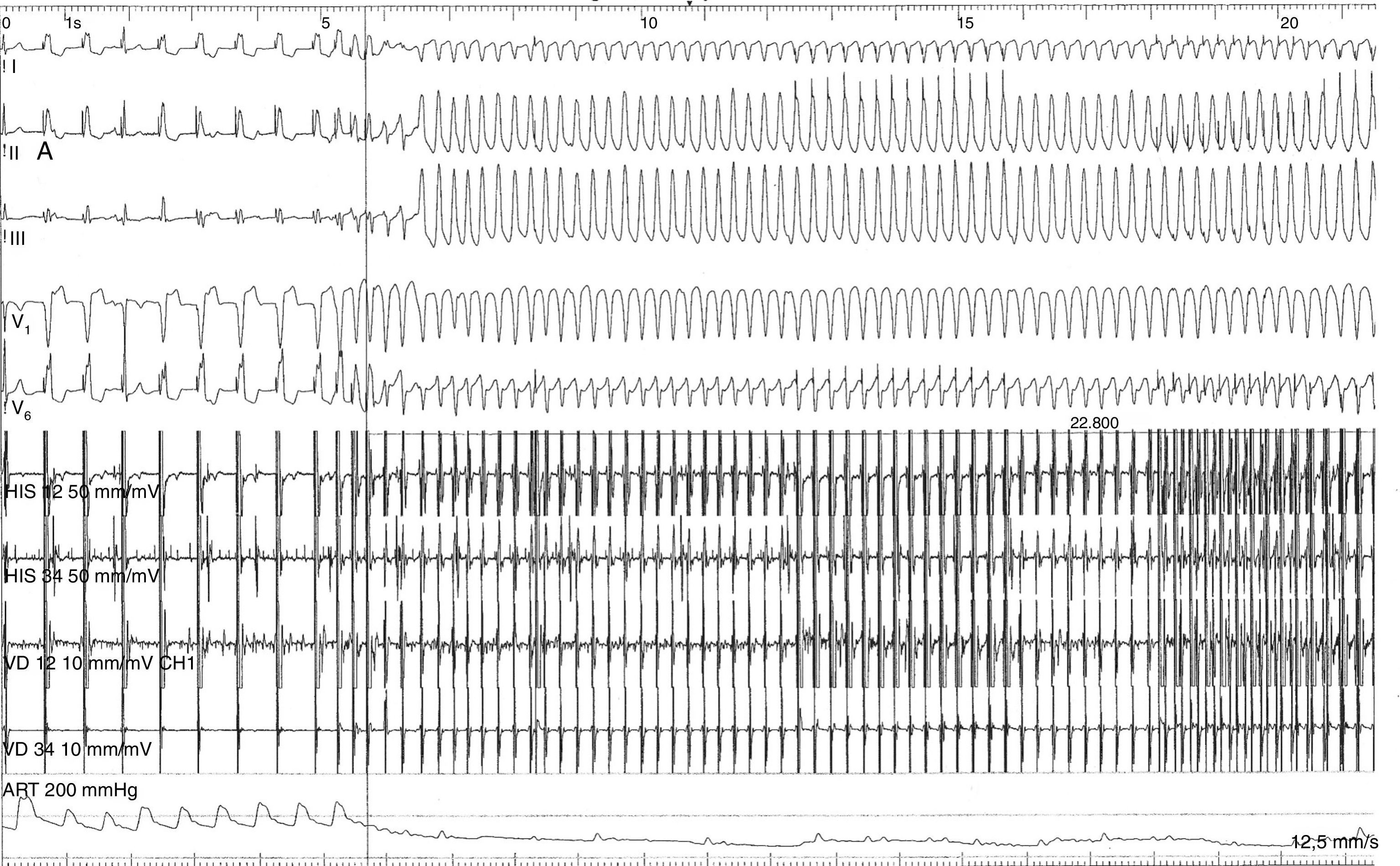
No sentido da estratificação do risco de morte súbita, foi efetuado o estudo eletrofisiológico (EEF), que documentou presença de inducibilidade de taquicardia ventricular monomorfa com morfologia de bloqueio completo do ramo esquerdo – eixo inferior, em duas ocasiões resultando em colapso hemodinâmico, numa carecendo de cardioversão elétrica por transformação polimorfa e noutra terminada após antitachycardia pacing (ATP) (Figura 4).
Foi decidida a implantação de cardioversor‐desfibrilhador implantável (CDI) monocâmara ventricular, que decorreu sem complicações.
As restantes medidas terapêuticas consistiram na restrição do esforço físico e na medicação em ambulatório com betabloqueador (bisoprolol 2,5mg qd), para além da medicação habitual do paciente.
O doente evoluiu assintomático e com boa tolerância à colocação do CDI. Está indicado o estudo genético e subsequente acompanhamento dos descendentes com eletrocardiografia e ecocardiografia1,2.
EpidemiologiaOs dados epidemiológicos relativos a esta patologia não são bem conhecidos. Estima‐se que a prevalência de DAVD na população geral varie entre 1/2000 e 1/5000, sendo os homens mais frequentemente atingidos do que as mulheres (3:1). A sua incidência varia de aproximadamente 1/1000 a 1/50000, dada a marcada variabilidade na distribuição geográfica3–5.
Sabe‐se que o diagnóstico de DAVD é feito, na grande maioria dos casos (80%), antes dos 40 anos. Mundialmente, em adultos jovens, é a causa de morte súbita cardíaca identificada em 5‐11% do total de casos. Num estudo oriundo do norte de Itália, a DAVD foi a principal causa de morte súbita em adultos jovens, particularmente atletas (22,4% do total de casos)6.
A DAVD deve, portanto, ser equacionada em indivíduos jovens que se apresentem pela primeira vez com história de arritmia, síncope ou paragem cardiorrespiratória, sendo atípico o episódio inaugural em idades mais avançadas. O diagnóstico tardio torna‐se ainda mais difícil devido à existência de possíveis fatores confundidores, como doença coronária concomitante7.
BiopatologiaDados da microscopia eletrónica8 apontam para alterações nas proteínas dos desmossomas como principal fator ultraestrutural que desencadeia falha na adesão dos miócitos, resultando em morte celular e substituição progressiva por tecido fibroadiposo, o que constitui o substrato anatomopatológico típico da DAVD. Estas modificações estruturais vão causar um stresse mecânico adicional ao miocárdio ventricular, com dilatação focal da cavidade, inicialmente localizada às áreas mais delgadas do VD (ápex, câmara de entrada e câmara de saída – triângulo da displasia) e, posteriormente, afetando toda a extensão do mesmo. De facto, este modelo pode explicar o surgimento, em fases mais precoces, de tropismo para o VD, mais distensível, menos espesso e mais assimétrico, comparativamente ao VE9.
No entanto, o envolvimento do VE é muito frequente (>50%) e a sua prevalência aumenta com a idade e o avançar da doença. Existem formas de apresentação em que o envolvimento do VE é o aspeto dominante, e a expressão final da patologia pode assemelhar‐se à da miocardiopatia dilatada10–12.
Na fase inicial da doença, as alterações estruturais estão confinadas ao referido triângulo da displasia e o doente é assintomático, podendo ainda assim estar em risco de morte súbita, particularmente durante o exercício físico. Posteriormente, as fibras miocárdicas contidas nas zonas de transformação fibroadiposa constituem o substrato para o desenvolvimento de arritmias ventriculares sintomáticas e as alterações funcionais no VD tornam‐se evidentes. A progressão da doença dá‐se do epicárdio para o endocárdio, sendo que, por fim, se instala difusamente por todo o VD, com dilatação e disfunção marcadas10.
No caso clínico apresentado, o episódio inaugural de síncope, num doente de 67 anos previamente assintomático, na presença das alterações estruturais e funcionais evidentes (dilatação marcada do VD com hipocinesia/acinesia segmentar e FE de 21%) é exemplificativo do possível caráter silencioso da DAVD, mesmo na presença de doença em fase avançada.
GenéticaTem existido grande variabilidade nos estudos relativamente à percentagem de casos hereditários de DAVD, com dados que variam entre os 30% e mais de 50%3,13.
As principais proteínas desmossomais cujas alterações se associam a DAVD são a placoglobina, desmoplaquina, placofilina‐2, desmogleína‐2 e a desmocolina‐2. Outros genes já foram identificados, como o da proteína TMEM43 (que se encontra associada a uma forma de alto risco de DAVD e que parece estar relacionada com a via lipogénica regulada pelo PPAR‐γ), o gene do TGF‐β3 e o gene do recetor rianodinico cardíaco (RYR2), cujos mecanismos patogénicos que levam a DAVD ainda se encontram em estudo. Encontram‐se por clarificar outras mutações que podem associar‐se ao fenótipo de DAVD1,14,15.
Até ao momento, foram identificados dois padrões de transmissão hereditária, formas autossómicas dominantes, mais comuns, de penetrância incompleta, e formas autossómicas recessivas, na qual a DAVD está integrada numa síndrome que inclui hiperqueratose palmoplantar e cabelo lanoso (síndromes de Naxos e Carvajal, associados aos genes da placoglobilina e desmoplaquina, respetivamente)16,17.
Devido ao padrão de penetrância incompleta, a idade de início dos sintomas e as manifestações clínicas podem variar dentro de indivíduos com a mesma mutação, sendo importante reconhecer os portadores assintomáticos em risco de vir a desenvolver a doença em determinado momento9,18.
História natural e manifestações clínicasA história natural da DAVD correlaciona‐se com o grau de progressão da disfunção ventricular direita e o desenvolvimento de sintomas.
A forma de apresentação inicial é vasta, desde palpitações, tonturas, mal‐estar geral, dor torácica e dispneia, até síncope e, menos frequentemente, morte súbita. O início dos sintomas ocorre geralmente entre a primeira e quinta décadas de vida, com idade média de diagnóstico aos 30 anos, sendo que raramente ocorre antes dos 12 e após os 60 anos.
A taxa de mortalidade global encontra‐se entre os 4‐20%, em ambos os sexos, com pico na quarta década de vida, e a taxa de morte súbita anual, em doentes com esta patologia, é de 1%. Devido a taxas de mortalidade elevadas, descritas em alguns estudos, e sabendo que a DAVD é uma doença de agravamento progressivo, é de extrema importância que esta condição seja identificada e o tratamento iniciado, prontamente3,9,15,19.
Um estudo realizado por análise macro e microscópica cardíaca, pós‐autópsia, revelou que em 1930 casos de morte súbita cardíaca, sem causa identificada, cerca de 10% mostravam evidência de DAVD, a maioria dos casos tendo ocorrido durante atividades sedentárias e apenas 3,5% durante a prática de exercício físico. No entanto, está bem demonstrado que, em determinadas populações, o exercício físico é uma importante causa de morte súbita em atletas20.
Como demonstrado, ainda que em muitos doentes esta entidade permaneça clinicamente silenciosa durante décadas, o risco de morte súbita cardíaca, ainda que relativamente baixo, é uma possibilidade que o clínico terá de ter em conta, o que leva a que a marcha diagnóstica deva ser célere, o que nem sempre sucede, especialmente nos casos esporádicos sem história familiar reconhecida21.
Abordagem diagnósticaNenhum método diagnóstico isolado será conclusivo e suficiente, particularmente no início da evolução da doença. Nesta fase, os dois meios complementares de diagnóstico essenciais, perante a suspeita de DAVD, são o electrocardiograma (ECG) e a ecocardiografia transtorácica (ETT)22.
EletrocardiogramaNa suspeita de DAVD, o ECG deverá fazer sempre parte da abordagem inicial do diagnóstico, sendo que alterações no ECG são observadas em cerca de 90% dos afetados4.
Apesar da sensibilidade do ECG inicial ser baixa (40‐50% no episódio inaugural), o acompanhamento a longo prazo com ECG seriados mostrou alta sensibilidade e especificidade. Os achados eletrocardiográficos mais importantes nos doentes com DAVD são as alterações da repolarização ventricular, sendo a inversão da onda T de V1‐V3 a mais frequente (54‐100% dos doentes). Esta alteração, apesar de fazer parte dos critérios major da Task Force 2010 para o diagnóstico de DAVD, não é específica desta entidade, podendo ser observada em indivíduos saudáveis. A onda épsilon é o achado mais específico de DAVD, está presente em 30% dos casos e corresponde ao atraso na ativação elétrica do VD. Outros achados descritos são: o padrão de bloqueio completo ou incompleto de ramo direito e o prolongamento da porção terminal do QRS23.
EcocardiografiaA ETT é o método mais vezes utilizado na deteção de alterações funcionais e estruturais do coração. É não‐invasivo e de fácil acesso na maioria dos centros hospitalares3,4,18.
Contudo, a posição retrosternal do VD e a complexidade da sua geometria dificultam a sua utilização como método de imagem isolado24.
As alterações mais comummente identificadas são: a dilatação do VD, particularmente da câmara de saída, podendo existir aneurismas regionais e aumento das aurículas; irregularidades morfológicas, que ocorrem em até 62% dos afetados, tais como, desorganização trabecular e banda moderadora hiperrefringente; diminuição da right ventricular fractional area change (RVFAC) que se correlaciona com a FE. Alterações da motilidade regional da parede do VD são observadas em 80% dos casos25. As medições ecocardiográficas atualmente incluídas nos critérios de diagnóstico da Task Force 2010 (ver abaixo) não são consideradas tão fidedignas, sendo menos sensíveis, do que as da RMC, principalmente devido a dificuldades na avaliação de transtornos regionais da motilidade da parede ventricular. Novas técnicas de ecocardiografia tridimensional propõem‐se ultrapassar algumas destas limitações. Alguns estudos demonstraram que além do valor diagnóstico da ecocardiografia, ela encerra igualmente um valor prognóstico na DAVD, tendo‐se verificado que a diminuição da TAPSE e da RVFAC estão associadas a episódios cardíacos major. No entanto, como sugerido anteriormente, eventos arrítmicos major podem ocorrer antes de existir disfunção sistólica, pelo que a estratificação de risco é dificultada pela grande variabilidade na expressão fenotípica da doença4.
Ressonância magnética cardíacaComo se verificou, a ecocardiografia nem sempre é suficiente para o diagnóstico de DAVD e a RM ultrapassa algumas das suas limitações. A RMC é um método não‐invasivo utilizado para detetar infiltração de tecido adiposo no miocárdio, estreitamento da parede e alterações da motilidade regional, pelo que os achados de RMC mais usados no diagnóstico são a presença de áreas hiperintensas na parede ventricular, que indicam deposição de gordura, dilatação da câmara de saída do VD, hipocinesia/acinesia/discinesia e dilatação do ventrículo e da aurícula direitos26.
Um método de RMC que parece ter boa correlação com a histopatologia, o grau de disfunção ventricular direita e a indução de taquicardia ventricular (TV) no EEF, é o realce tardio após injeção de contraste paramagnético, usado para detetar tecido fibroso. Esta técnica mostrou positividade em 67% dos doentes com DAVD27.
Alguns estudos mostraram que, na DAVD, a RMC tem um valor diagnóstico superior à ecocardiografia convencional22,28. No entanto, também está sujeita a falsos positivos, principalmente se forem utilizados critérios estritamente com base nas alterações fibroadiposas e da espessura da parede ventricular18,24,29.
Para além da percentagem de falsos positivos, algumas limitações da RMC prendem‐se com a variabilidade interobservador na sua interpretação, a existência de artefactos relacionados com fenómenos arritmogénicos, o uso limitado na presença de dispositivos intracardíacos, o custo e a acessibilidade em centros hospitalares menos diferenciados30.
Eletrocardiograma dinâmico de 24 horasVários estudos demonstraram a importância do ECGD/Holter 24 horas na DAVD, quer na identificação de sístoles prematuras ventriculares, quer na deteção de arritmias ventriculares potencialmente fatais, tendo sido evidenciada uma correlação entre as duas31–33.
Estudo eletrofisiológicoExistem dados contraditórios no que concerne à estimulação ventricular programada e indução de taquicardia ventricular na estratificação de risco em indivíduos com DAVD. No entanto, a indução de taquicardia ventricular, apesar de não ser fator preditivo de eventos arrítmicos futuros pode identificar os doentes com maior risco de progressão da doença e de morte súbita34.
Num estudo de follow‐up de 62 doentes com diagnóstico de DAVD pela TF 2010, 55% apresentaram indutibilidade de TV monomórfica, sendo que, após dez anos de observação, estes doentes apresentaram maior frequência de morte de causa cardíaca, transplante cardíaco, fibrilhação ventricular e TV com compromisso hemodinâmico18,35.
A Task Force de consenso internacional de julho 2015 recomenda o EEF no diagnóstico e/ou avaliação de casos suspeitos de DAVD (classe IIa)36.
Testes genéticosNão se preconiza a utilização de testes genéticos em todos os doentes com suspeita de DAVD. Particularmente, em doentes cujo diagnóstico definitivo foi feito com base nos critérios da TF 2010 o estudo genético não é uma necessidade, podendo ser útil quando for duvidosa a valorização dos achados clínicos, eletrocardiográficos e imagiológicos, mas o diagnóstico for possível (classe IIb)37.
Com a identificação do gene da proteína TMEM43, anteriormente mencionada, abre‐se uma nova janela de adequabilidade para os testes genéticos na estratificação de risco, pois esta parece estar associada a uma mortalidade de 50% em homens até aos 39 anos de idade38.
Diagnóstico e estratificação de riscoAs principais causas para o diagnóstico incorreto de DAVD são a interpretação incorreta da RMC e o desconhecimento dos critérios da TF 2010. Neste caso clínico, o diagnóstico de DAVD foi feito segundo esses critérios, que reviu os anteriores datados de 1994. Estes tomam em linha de conta aspetos estruturais e funcionais do VD, aspetos histopatológicos, anomalias de repolarização e despolarização no ECG, presença de arritmias e história familiar1.
Segundo os mesmos, o caso aqui discutido apresentava dois critérios major e um minor, pelo que o diagnóstico definitivo foi assumido.
Major: por RMC – acinesia do VD e FE≤40%; por ECG – ondas T invertidas nas derivações pré‐cordiais direitas (V1‐3).
Minor: no ECGD/Holter –>500 extrassístoles ventriculares nas 24 horas.
Um estudo de performance das técnicas de diagnóstico imagiológico observou que apenas 50% dos pacientes com diagnóstico de DAVD por RMC cumpriam os critérios ecocardiográficos da TF 2010, o que evidencia a importância da RMC na abordagem desta patologia39.
A busca por indicadores de risco que permitam a estratificação do mesmo mostrou, até à data, que a disfunção VD e VE, presença de mutação do gene da TMEM43, inversão da onda T nas derivações à esquerda de V3 e a presença de taquicardia ventricular estabelecida, são as mais convincentes. Dado o caráter progressivo da doença, a estratificação de risco deverá ser considerada um processo dinâmico sujeito a reavaliação temporal34.
Abordagem terapêuticaA estratégia terapêutica terá de ser sempre direcionada primariamente para a prevenção de morte súbita cardíaca, sendo que a melhor forma de prevenir este desfecho dramático ainda não está bem definida.
Pelos motivos adiantados anteriormente, a restrição do exercício físico terá de ser uma realidade nos doentes afetados com DAVD, pelo que os mesmos não deverão participar, por norma, em desportos de alta competição (classe Ic)36,40,41.
Fármacos antiarrítmicosRelativamente aos betabloqueadores, apesar dos estudos específicos para DAVD serem escassos, a sua importância pode ser induzida a partir do conhecimento do efeito protetor de risco de morte súbita cardíaca em doentes sem DAVD e a sua utilização é recomendada (classe Ic). Com base na análise dos efeitos do exercício físico sobre o miocárdio dos doentes afetados, o mecanismo de atuação destes fármacos passará pela frenação dos conhecidos efeitos nocivos do sistema nervoso simpático nesta patologia.
Um estudo demonstrou maior eficácia da amiodarona na prevenção de arritmias ventriculares clinicamente significativas e associou o sotalol a um risco aumentado dos mesmos fenómenos42. Outro estudo mostrou superioridade do sotalol face à amiodarona, e apontou para a possível eficácia do verapamil e dos betabloqueadores43. Deverão ser efetuados mais estudos que comprovem a eficácia destes e outros fármacos na prevenção primária em portadores de gene relacionado com a doença.
Devido aos dados contraditórios presentes na literatura, no caso clínico apresentado optou‐se por uma dose baixa de betabloqueador como coadjuvante do CDI36,42,43.
Ablação por radiofrequênciaDevido ao caráter segmentar e à natureza progressiva da DAVD, a ablação por radiofrequência não constitui um tratamento definitivo e não deverá ser utilizada isoladamente ou como primeira linha. Alguns casos em que o foco arritmogénico é bem localizado poderão beneficiar desta técnica44.
Cardioversor‐desfibrilhador implantávelAs recomendações atuais são para a colocação de CDI em todos os doentes que cumpram os critérios da TF 2010, particularmente na presença de história familiar de morte súbita cardíaca, taquicardia ventricular sustentada ou síncope recente inexplicada. O número de sístoles prematuras ventriculares mostrou ser proporcional à adequabilidade do CDI e à frequência dos eventos arrítmicos. A inducibilidade no EEF mostrou ser preditora de adequabilidade do CDI9,12.
Como prevenção primária, o CDI deverá ser opção em doentes de alto risco, apesar de não existir consenso nos seus determinantes. A síncope mostrou uma relação preditiva com intervenções adequadas do CDI em doentes com implantação profilática do dispositivo, designadamente na prevenção de eventos potencialmente fatais45,46.
Como prevenção secundária, o consenso é total e deverá ser opção na presença de TV ou FV documentadas45,47,48.
As principais complicações relacionadas com a implantação de CDI incluem hematoma da loca, problemas relacionados com a colocação de elétrodos, bem como derrame e infeção pericárdicos. Especificamente, nos doentes com DAVD, pode existir perfuração da parede do VD, alterações estruturais que prejudiquem a colocação dos elétrodos do CDI, e interfiram com a sensibilidade e o ritmo cardíacos. Além disso, em indivíduos jovens, o CDI necessitará de ser substituído e os elétrodos recolocados futuramente49.
No presente caso clínico, dada a presença de síncope de origem indeterminada com indução de taquicardia ventricular monomorfa e consequente colapso hemodinâmico, foi decidida a implantação de CDI tal com indicam as recomendações (classe Ic)47.
Acompanhamento dos familiaresOs estudos genéticos devem ser utilizados na deteção e estratificação do risco, em familiares assintomáticos dos doentes; no entanto, a presença de mutação não garante que existirá tradução clínica, apenas indica a presença de risco.
A única indicação formal para testes genéticos em familiares será quando se identifica uma mutação causadora num caso índice de DAVD.
É de salientar que alguns estudos demonstraram que, em casos de doença familiar, muitos membros inicialmente sem doença objetivável acabaram por desenvolvê‐la numa idade mais avançada (50%)2,40,50.
O diagnóstico de DAVD nos familiares de um doente afetado baseia‐se na presença de inversão da onda T nas derivações V1‐V3, em indivíduos com mais de 14 anos (é uma alteração observada em jovens até esta idade, saudáveis), potenciais tardios por signal‐averaged ECG (SAECG), taquicardia ventricular com padrão de bloqueio de ramo esquerdo no ECG, no ECGD/Holter 24 horas ou durante a prova de esforço, mais de 200 extrassístoles no ECGD/Holter 24 horas e dilatação ligeira ou redução da FE do VD e hipocinesia regional1.
ConclusãoApesar de cada vez mais se preconizar a utilização da nomenclatura miocardiopatia arritmogénica no lugar de DAVD51, muito por força do conhecimento crescente de que o envolvimento do VE pode ser inclusivamente o aspeto predominante, este envolvimento não se encontra ainda contemplado nos critérios de diagnóstico da TF 2010.
Como tal, e dado que no presente caso clínico o VE se encontrava preservado, optou‐se por mencionar a entidade pelo seu nome clássico no corpo de texto.
A relevância do caso clínico apresentado prende‐se com diversos aspetos que vêm ao encontro dos dados apresentados na literatura. Em primeiro lugar, exemplifica o caráter clinicamente silencioso da DAVD e a possibilidade de apresentação tardia da mesma, bem como a possível existência de um grau de deformação estrutural e de compromisso da função ventricular direita elevados, representativos de uma fase histopatológica avançada, num doente previamente assintomático.
Sintomas como palpitações e síncope podem ser a manifestação de arritmias ventriculares, que podem ir desde extrassístoles ventriculares frequentes, até taquicardia ventricular mantida, sendo que a frequência dessas manifestações arrítmicas é proporcional à gravidade da doença.
A distribuição preferencial, aparentemente circunscrita a determinados subgrupos populacionais, poderá relacionar‐se com fatores genéticos. No entanto, com maior probabilidade dever‐se‐á a dificuldades diagnósticas inerentes à própria patologia, pelo que a prevalência real da mesma tenderá a ser superior19.
Neste doente, sem história familiar relevante, colocou‐se também a dificuldade diagnóstica trazida por fatores confundidores, clinicamente legítimos e epidemiologicamente mais relevantes, como a suspeita de síndrome coronária aguda e tromboembolismo pulmonar, que levaram à execução de vários exames complementares de diagnóstico, comportando para o doente uma sobrecarga emocional e financeira marcada. A importância do ECG e do ecocardiograma iniciais ficaram bem patentes, assim como as limitações desta última técnica na aferição de distúrbios da motilidade concernentes ao VD.
A RM evidenciou a sua superioridade em relação ao ecocardiograma e, neste caso, foi o exame decisivo para o diagnóstico desta entidade, independentemente da presença de tecido adiposo ou de adelgaçamento da parede ventricular, que não foram descritos neste doente.
É de salientar a importância do EEF na documentação da taquicardia ventricular polimorfa que levou diretamente à decisão de colocação do CDI, particularmente após o ECGD/Holter 24 horas e a prova de esforço não identificarem a presença de quaisquer arritmias ventriculares.
Foi identificada uma relação entre o exercício físico e a aceleração da progressão da doença52, o que apoia o modelo que relaciona o stresse mecânico com esta miocardiopatia, explica a apresentação precoce e mais grave em atletas, e favorece a indicação para restrição do exercício físico nos doentes afetados.
Numa era pós‐sequenciação do genoma humano, um grande número de doentes portadores de genes de risco para DAVD continua por identificar. São indivíduos assintomáticos que poderão ter alterações estruturais em várias fases de progressão da doença. Para as famílias sem mutação identificada, as dificuldades no diagnóstico inicial desta entidade clínica deverão constituir razão de preocupação, sabendo que a morte súbita pode ser a primeira manifestação da doença.
A frequência de arritmias malignas como primeiro sinal de DAVD evidencia a importância da sensibilidade clínica a sinais e sintomas que possam sugerir o diagnóstico e a importância do rastreio eletrocardiográfico e imagiológico precoces.
Infelizmente, ainda não existe um algoritmo de risco credível para predizer a probabilidade de um indivíduo desenvolver uma arritmia fatal que, desta forma, oriente definitivamente a decisão de implantação de CDI profilático.
Assim, apesar de as indicações para colocação de CDI não reunirem consenso, este continua a ser a única terapêutica que demonstrou verdadeiramente reduzir o risco de morte súbita nestes doentes. Se bem que alguns estudos apontam para a eficácia do tratamento apenas com fármacos antiarrítmicos em doentes com arritmias cardíacas hemodinamicamente estáveis, as guidelines atuais promovem a prevenção secundária com colocação de CDI em doentes com taquicardia ou fibrilhação ventricular, e como prevenção primária em doentes de alto risco (idade jovem, atletas, história familiar pesada e episódios frequentes de síncope)45,48.
Os objetivos futuros passarão por atrasar a progressão da doença ou mesmo revertê‐la.
Responsabilidades éticasProteção de pessoas e animaisOs autores declaram que para esta investigação não se realizaram experiências em seres humanos e/ou animais.
Confidencialidade dos dadosOs autores declaram ter seguido os protocolos do seu centro de trabalho acerca da publicação dos dados de pacientes.
Direito à privacidade e consentimento escritoOs autores declaram ter recebido consentimento escrito dos pacientes e/ou sujeitos mencionados no artigo. O autor para correspondência deve estar na posse deste documento.
Conflito de interessesOs autores declaram não haver conflito de interesses.
