





A miocardiopatia arritmogénica do ventrículo direito – também denominada de displasia arritmogénica do ventrículo direito – é uma patologia em que se verifica a substituição do miocárdio por tecido fibroso ou fibroadiposo predominantemente no ventrículo direito e que se caracteriza clinicamente por arritmias ventriculares potencialmente letais, sendo uma das causas mais relevantes de morte súbita cardíaca. A sua prevalência exata é desconhecida, no entanto, estima‐se que seja de cerca de 1: 5.000 na população adulta. O diagnóstico pode ser efetuado mediante a constatação de alterações estruturais e funcionais do ventrículo direito, alterações eletrocardiográficas – da condução em ECG basal, arritmias ventriculares – e da história familiar. A existência de critérios de diagnóstico facilita o reconhecimento e interpretação das características clínicas não específicas desta entidade.
Apresenta‐se um caso clínico em que o diagnóstico de miocardiopatia arritmogénica do ventrículo direito foi desencadeado a partir da suspeita de patologia ventricular direita suscitada por ecocardiograma transtorácico. A análise do ECG serviu para o confirmar, tendo‐se detetado ondas épsilon que em geral passam despercebidas, mas que neste caso foram a chave para o diagnóstico tendo‐se também aferido a sua existência em técnicas de ECG não convencional como o ECG modificado de Fontaine.
A evolução subsequente do quadro clínico culminou com a ocorrência de taquicardia ventricular o que motivou a implantação de cardiodesfibrilhador implantável (CDI).
Arrhythmogenic right ventricular cardiomyopathy, also known as arrhythmogenic right ventricular dysplasia, is a condition in which myocardium is replaced by fibrous or fibrofatty tissue, predominantly in the right ventricle. It is clinically characterized by potentially lethal ventricular arrhythmias, and is a leading cause of sudden cardiac death. Its prevalence is not known exactly but is estimated at approximately 1:5000 in the adult population. Diagnosis can be on the basis of structural and functional alterations of the right ventricle, electrocardiographic abnormalities (including depolarization and repolarization alterations and ventricular arrhythmias) and family history. Diagnostic criteria facilitate the recognition and interpretation of non‐specific clinical features of this disease.
The authors present a case in which the diagnosis of arrhythmogenic right ventricular cardiomyopathy was prompted by the suspicion of right ventricular disease on transthoracic echocardiography. This was confirmed by detection of epsilon waves on analysis of the ECG, which generally go unnoticed but in this case were the key to the diagnosis. Their presence was also shown by non‐conventional ECG techniques such as modified Fontaine ECG.
The course of the disease culminated in the occurrence of ventricular tachycardia, which prompted placement of an implantable cardioverter‐defibrillator.
Mulher de 46 anos, referenciada a consulta de cardiologia devido a dilatação das cavidades direitas detetada em ecocardiograma transtorácico de rotina. Assintomática. A doente não referia antecedentes pessoais conhecidos de doença cardíaca, mas referia história familiar de uma filha com diagnóstico de miocardiopatia arritmogénica do ventrículo direito (MAVD), com atingimento ventricular esquerdo, e portadora de cardiodesfibrilhador implantável (CDI), seguida noutra instituição hospitalar e outra filha sem doença. Ao exame objetivo não apresentava quaisquer alterações.
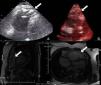
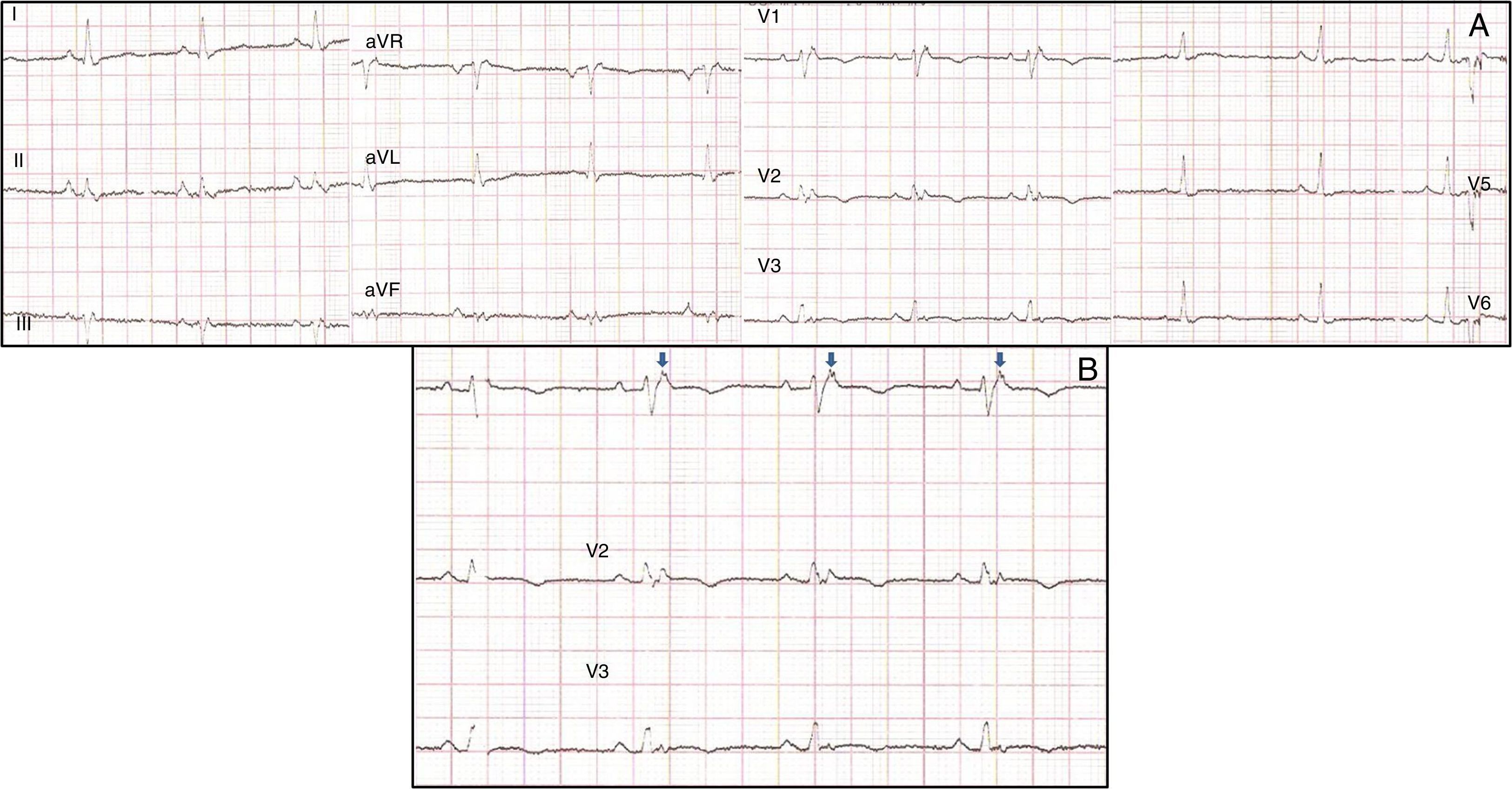
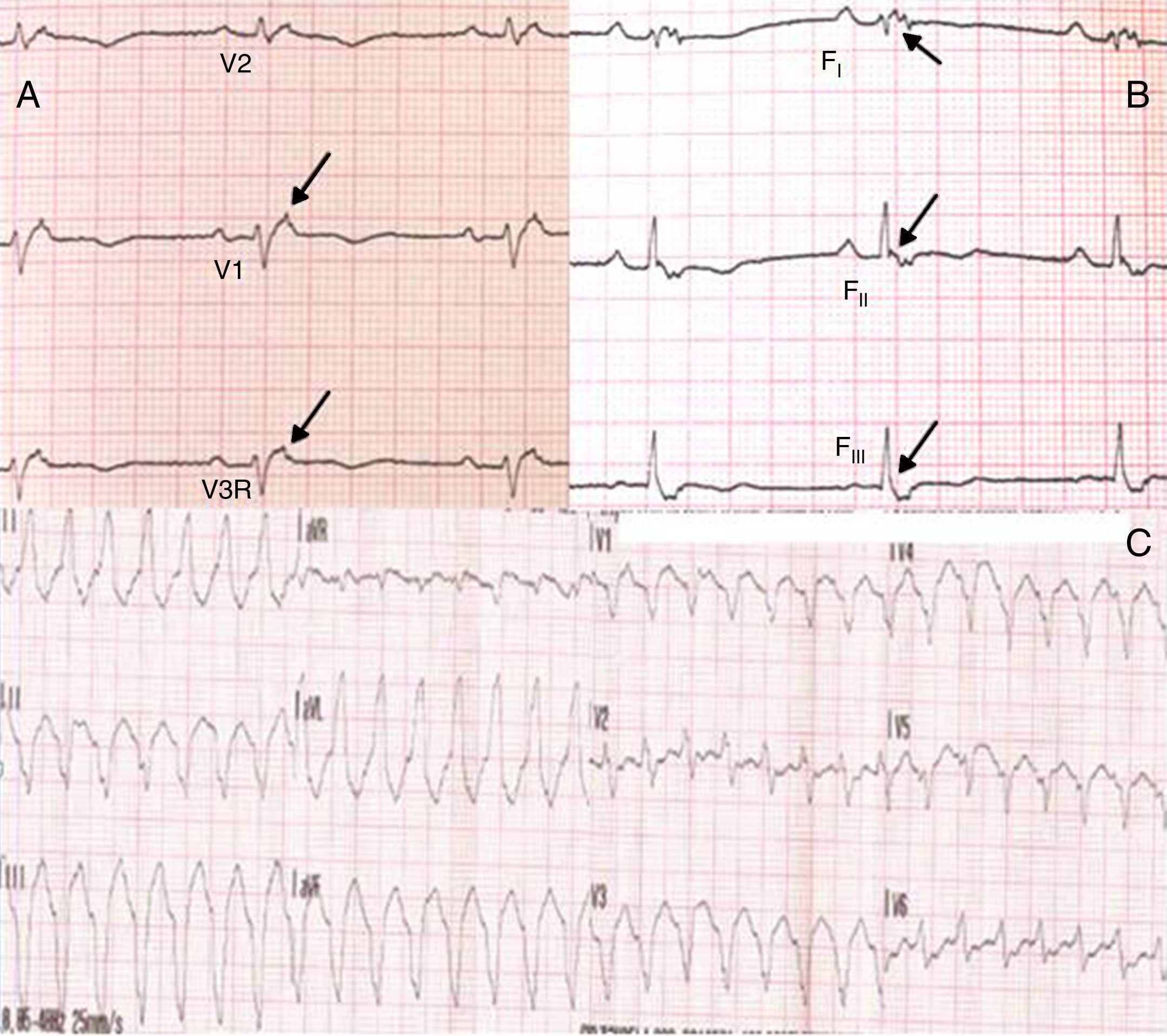
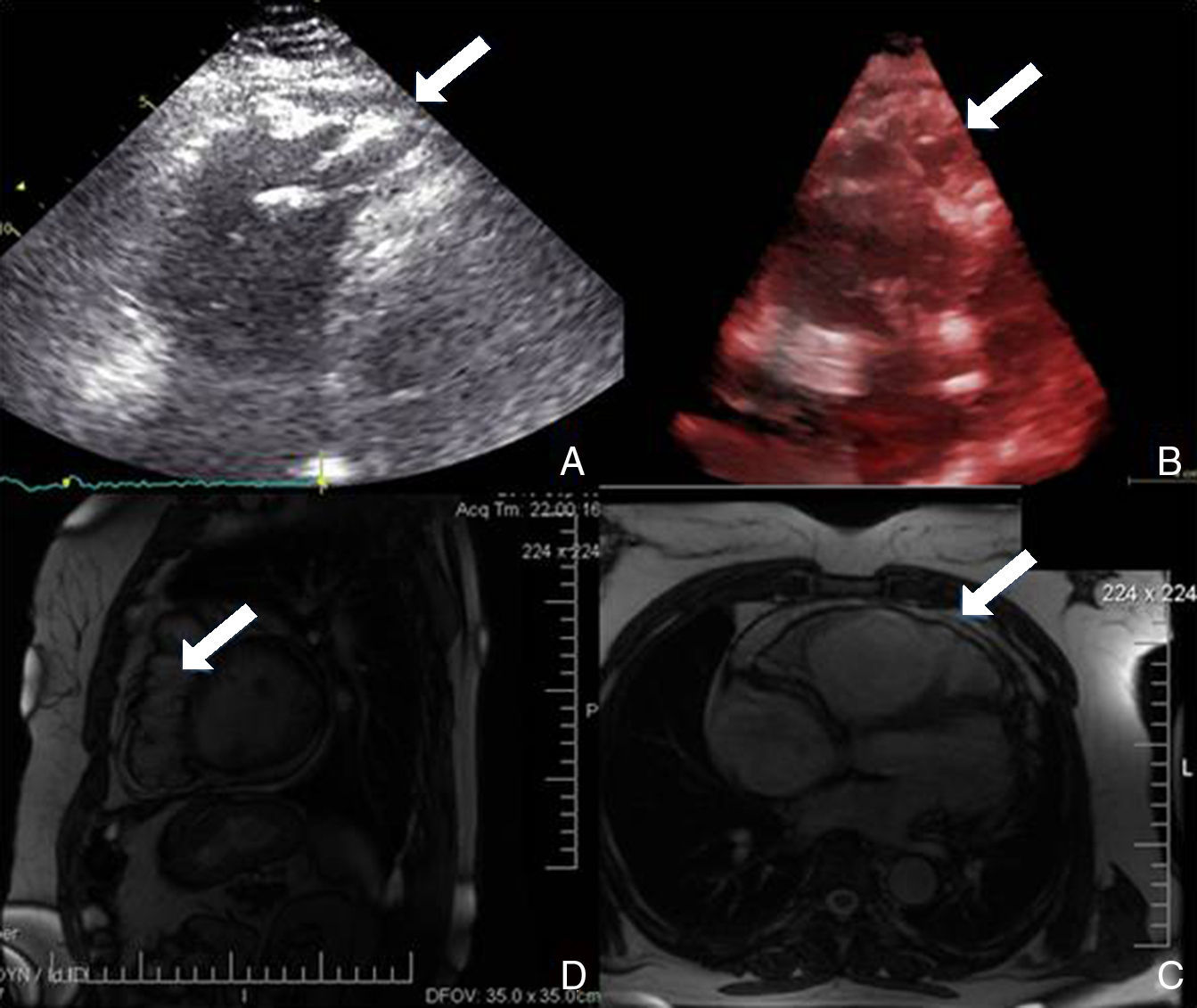
No âmbito da investigação diagnóstica foi realizado eletrocardiograma (ECG) de 12 derivações, que mostrou padrão de bloqueio de ramo direito, conjuntamente com a presença de onda épsilon e inversão das ondas T nas derivações V1 a V3 (Figura 1). Na caracterização das alterações eletrocardiográficas da MAVD foi realizado ECG das derivações precordiais direitas e ECG modificado de Fontaine, este último mediante a seguinte disposição de elétrodos: elétrodo do membro superior direito sobre a área do manúbrio do esterno, elétrodo do membro superior esquerdo sobre a área da apófise xifoide e elétrodo do membro inferior esquerdo na área correspondente de V4, tendo‐se mantido a velocidade de registo a 25mm/s e voltagem a 10mm/mV (Figura 2). Ambos os métodos mostraram uma nítida clarificação das ondas épsilon, em especial neste último (Figura 3 A e B). Procedeu‐se à realização de ecocardiograma transtorácico bidimensional e a três dimensões na nossa instituição hospitalar que mostrou ventrículo direito severamente dilatado e hipocinético, com presença de trabéculas profundas e vários tendões no ápice e dilatações saculiformes na parede livre do ventrículo (Figura 4A e 4B); a ressonância magnética nuclear cardíaca (RMN‐C) mostrou a existência de pequenos focos de infiltração adiposa subepicárdica na parede livre e inferior do ventrículo direito, septo interventricular e parede livre do ventrículo esquerdo; com realce tardio observou‐se a existência de focos de realce intramiocárdico no septo interventricular compatíveis com fibrose (Figura 4C e D).
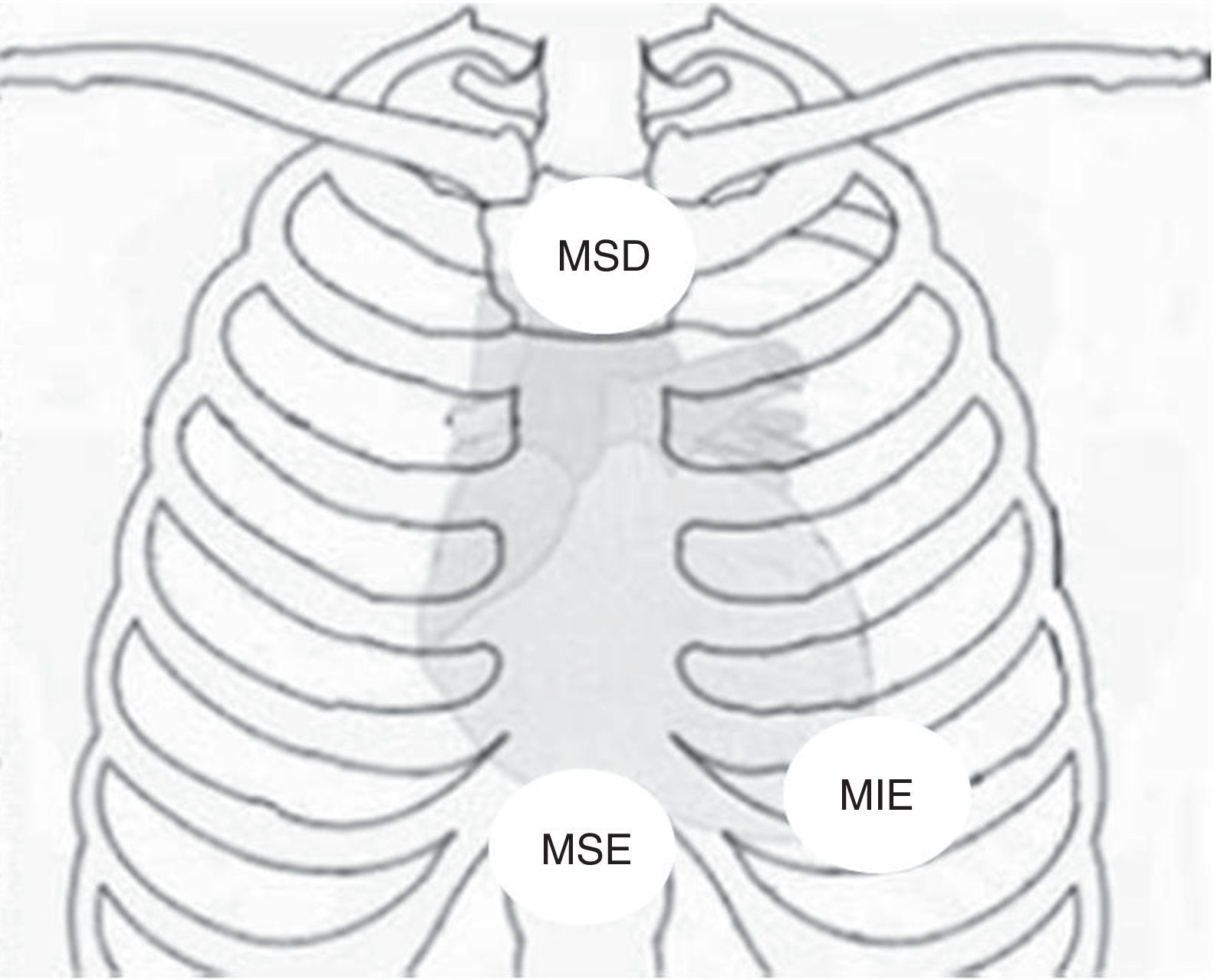
Disposição dos elétrodos no ECG de Fontaine. MIE: membro inferior esquerdo; MSD: membro superior direito; MSE: membro superior esquerdo. Fonte: Chiladakis et al.8.
O estudo de mutações dos genes codificantes dos componentes dos desmossomas placofilina‐2 (PKP2), desmogleína‐2 (DSG2) e desmoplaquina (DSP) encontrou uma mutação non‐sense no exão 12 do gene PKP2, também presente na filha afetada. Esta mutação ocorre em 11‐43% dos doentes com MAVD.
Pelo facto de a doente se manter assintomática e no Holter de 24h apenas se verificar a existência de extrassistolia ventricular em pares e triplet foi instituída terapêutica com amiodarona 200mg id e prosseguiu seguimento clínico. Cerca de um ano depois, aquando de episódio de lipotimia, foi constatada a existência de taquicardia ventricular (TV) não sustentada, com configuração de bloqueio de ramo esquerdo e eixo superior (Figura 3C). Esta situação, para além de também constituir critério major para o diagnóstico desta entidade, motivou a implantação de CDI; cerca de um mês após a implantação verificou‐se episódio de TV que converteu a ritmo sinusal com aplicação de choque apropriado após insucesso de pacing antitaquicardia. Desde este episódio e decorridos cerca de 18 meses não se verificou a recorrência de disritmias ventriculares.
DiscussãoA MAVD é uma entidade clínica caracterizada por arritmias ventriculares e patologia ventricular que do ponto de vista macroscópico se caracteriza pela substituição do miocárdio por tecido fibroso ou fibroadiposo, predominantemente no ventrículo direito, mas que pode afetar o ventrículo esquerdo1,2.
A incidência estimada é de 1: 5.000 na população2; é uma causa relevante de morte súbita cardíaca (MSC), com uma incidência estimada entre 0,08‐9%1. No entanto, em jovens adultos pode contribuir com até 10,8% dos casos de MSC3.
A apresentação clínica mais comum ocorre entre os 10‐50 anos de idade, com idade média de diagnóstico por volta dos 30 anos4. Os principais sintomas de apresentação são tonturas, palpitações e síncope4; no entanto, a maioria dos pacientes é assintomática e a suspeita diagnóstica surge na sequência de alterações eletrocardiográficas inespecíficas, alterações ecocardiográficas ou na documentação de arritmias ventriculares4.
Estima‐se que cerca de 30% dos casos têm agregação familiar. Os dois padrões de hereditariedade são o autossómico dominante, mais comum, e o autossómico recessivo, denominado de doença de Naxos, na qual a MAVD é parte duma síndrome cardiocutânea que inclui hiperqueratose palmo‐plantar e cabelo lanudo.
A MAVD deve ser considerada em pacientes que se apresentam com TV sintomática ou assintomática, com configuração do tipo bloqueio de ramo esquerdo na ausência de cardiopatia aparente5,6. O diagnóstico definitivo requer documentação histológica da substituição do miocárdio por tecido fibroadiposo no ventrículo direito; no entanto, este critério não é prático em contexto clínico, pelo que outros dados são utilizados para esse fim. Em 1994 foi elaborado o primeiro documento com os critérios diagnósticos desta entidade tendo este consenso sido revisto em 20106, conforme consta na Tabela 1. O diagnóstico definitivo é realizado na presença de dois critérios major ou de um critério major e dois minor ou de quatro critérios minor de diferentes categorias. O diagnóstico borderline requer um critério major e um minor ou três critérios minor de diferentes categorias. O diagnóstico possível requer um critério major ou dois minor de diferentes categorias. Neste caso, o cumprimento dos critérios de história familiar positiva para MAVD e de constatação de ondas épsilon no ECG de 12 derivações tornou‐se suficiente para o diagnóstico; este achado eletrocardiográfico encontra‐se presente em 30% dos doentes com MAVD e representa potenciais de baixa amplitude causados pela ativação tardia de algumas porções do ventrículo direito. São tipicamente mais bem identificadas nas derivações V1 a V3, assim como nas derivações precordiais direitas. A duplicação da sensibilidade do registo e da utilização das definições do filtro de 40Hz em vez de 150Hz de modo a diminuir o ruído aumentam a capacidade da sua deteção7. No entanto, a sua presença não é específica da MAVD podendo estar presentes aquando da existência de alterações do ventrículo direito decorrentes de enfarte agudo do miocárdio ou sarcoidose cardíaca7. A utilidade do ECG de Fontaine reside na maior capacidade de demonstração da existência das ondas épsilon relativamente ao ECG standard de 12 derivações e no ECG das derivações precordiais direitas8; de acordo com Wang et al. a utilização do ECG modificado de Fontaine permite duplicar ou triplicar a percentagem de deteção de ondas épsilon relativamente ao ECG standard de 12 derivações8; uma explicação para este facto pode residir no facto do ECG de Fontaine registar os potenciais com origem no ventrículo direito, desde o infundíbulo à área do diafragma8. Por conseguinte, este método é particularmente útil na abordagem aos casos suspeitos de MAVD, principalmente na presença de bloqueio completo do ramo direito do feixe de His, nos quais o reconhecimento das ondas épsilon é ainda mais relevante dado o facto de a duração de QRS >110ms não permitir a aferição de vários critérios eletrocardiográficos potencialmente úteis para o diagnóstico desta entidade7. Apesar de neste caso clínico o ECG de Fontaine não ser decisivo para a sua constatação, o interesse da sua realização reside nesta sua capacidade e também do facto de constituir um método eletrocardiográfico muito simples e relativamente desconhecido.
Critérios revistos da Task Force em 2010 para o diagnóstico da miocardiopatia arritmogénica do ventrículo direito (MAVD)
| Critérios revistos da Task Force (2010) | |
| Disfunção global ou regional e alterações estruturais | |
| Major | Em ecocardiograma 2D:‐ Acinésia regional do VD, discinésia ou aneurisma‐ E um dos seguintes (telediastólico):--‐ CSVD PLAX ≥32mm (corrigido para a área de superfície corporal [PLAX/ASC] ≥19mm/m2)--‐ CSVD PSAX ≥36mm (corrigido para a área de superfície corporal [PLAX/ASC] ≥21mm/m2)--‐ Ou alteração fracional da área ≤33% |
| Por RMN‐ Acinésia regional do VD, discinésia ou dissincronia na contração do VD‐ E um dos seguintes:-- Quociente do volume telediastólico do VD e ASC ≥110ml/m2 (homem) ou ASC ≥100ml/m2 (mulher)-- Ou fração de ejeção do VD ≤40% | |
| Por angiografia do VD‐ Acinésia regional do VD, discinésia ou aneurisma | |
| Minor | Em ecocardiograma 2D:‐ Acinésia regional do VD ou discinésia‐ E um dos seguintes (telediastólico):--‐ CSVD PLAX ≥29mm e <32mm (corrigido para a área de superfície corporal [PLAX/ASC] ≥16mm/m2 e <19mm/m2)--‐ CSVD PSAX ≥32mm e <36mm (corrigido para a área de superfície corporal [PLAX/ASC] ≥18mm/m2 e <21mm/m2)--‐ Ou alteração fracional da área >33% e ≤40% |
| Por RMN‐ Acinésia regional do VD ou discinésia ou dissincronia na contração do VD‐ E um dos seguintes:-- Quociente do volume telediastólico do VD e ASC ≥100 e <110ml/m2 (homem) ou ≥90 e <100ml/m2 (mulher)-- Ou fração de ejeção do VD >40% e ≤45% | |
| Caracterização histológica da parede ventricular | |
| Major | Miócitos residuais <60% em análise morfométrica (ou <50% por estimativa) com substituição fibrosa da parede livre do VD em mais de uma amostra com ou sem substituição adiposa na biópsia miocárdica |
| Minor | Miócitos residuais 60‐75% por análise morfométrica (ou de 50‐65% por estimativa) com substituição fibrosa da parede livre do VD em mais de uma amostra com ou sem substituição adiposa na biópsia miocárdica |
| Alterações da repolarização ventricular | |
| Major | ‐ Ondas T invertidas em V1, V2 ou V3 em indivíduos >14 anos de idade na ausência de bloqueio completo de ramo direito com QRS ≥120ms |
| Minor | ‐ Ondas T invertidas em V1 e V2 em indivíduos >14 anos de idade na ausência de bloqueio completo de ramo direito com QRS ≥120ms ou em V4, V5 ou V6‐ Ondas T invertidas em V1, V2, V3 e V4 em indivíduos >14 anos na presença de bloqueio completo de ramo direito com QRS ≥120ms |
| Alterações da condução/despolarização | |
| Major | ‐ Onda épsilon nas derivações de V1 a V3 |
| Minor | ‐ Potenciais tardios no ECG de alta resolução em mais do que um dos três seguintes parâmetros na ausência de QRS ≥ 110 ms no ECG standard de 12 derivações:--‐ Duração do QRS filtrado (fQRS) ≥114ms--‐ Duração do QRS terminal <40μV (duração de sinal de baixa amplitude) ≥38ms--‐ Raiz média quadrática dos potenciais nos 40ms terminais da ativação ventricular (RMS40 [mV]) ≤20μV‐ Duração da ativação terminal do QRS ≥ 55 ms medida do nadir da onda S ao término do QRS, incluindo R’ em V1, V2, V3 na ausência de bloqueio completo de ramo direito do feixe de His |
| Arritmias | |
| Major | Taquicardia ventricular sustentada ou não sustentada com morfologia de bloqueio completo do ramo esquerdo com eixo superior (QRS negativo ou indeterminado em II, III, aVF e positivo em aVL) |
| Minor | Taquicardia ventricular sustentada ou não sustentada com configuração de câmara de saída do VD, morfologia de bloqueio completo do ramo esquerdo com eixo inferior (QRS positivo em II, III e aVF e negativo em aVL) ou de eixo indeterminado> 500 extrassístoles ventriculares no registo Holter de 24 h |
| História familiar | |
| Major | ‐ MAVD confirmada em familiar do 1.° grau que cumpre os critérios da Task Force‐ MAVD confirmada por histopatologia na autópsia ou cirurgia em familiar do 1.° grau‐ Identificação de mutação patogénica categorizada como associada ou provavelmente associada a MAVD* em doente sob avaliação |
| Minor | ‐ História de MAVD em familiar do 1.° grau no qual não é possível ou a exequibilidade de confirmação de presença de critérios da Task Force é difícil‐ Morte súbita cardíaca (idade <35 anos) devido a suspeita de MAVD em familiar de 1.° grau‐ MAVD confirmada por histopatologia ou pelos critérios da presente Task Force em familiar do 2.° grau |
ASC: área de superfície corporal; CSVD: câmara de saída do ventrículo direito; PLAX: paraesternal longo eixo; PSAX: paraesternal curto eixo.
* Por mutação patogénica entende‐se uma alteração de ADN associada a MAVD que altera ou que é expectável que altere uma proteína codificada, não é observada ou é rara numa população de controlo sem MAVD e que altera ou é previsível que altere a função ou a estrutura de proteína ou que tenha demonstrado associação a fenótipo da patologia numa árvore familiar.
Adaptado a partir de 2010 Revised Task Force criteria for the diagnosis of Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC), disponível no artigo Clinical manifestations and diagnosis of arrhythmogenic right ventricular cardiomyopathy, disponível em www.uptodate.com, acedido em 01 de Outubro de 2013.
A realização de ressonância magnética cardíaca nesta situação – e após a realização de ecocardiograma transtorácico na nossa instituição hospitalar – não era obrigatória. No entanto, dada a sua exequibilidade e a existência de possibilidade de constatação das características imagiológicas desta miocardiopatia procedeu‐se à sua realização9.
A utilidade do teste genético permanece como alvo de debate. Embora as mutações dos genes codificantes dos componentes dos desmossomas sejam relativamente frequentes nos doentes sob avaliação de MAVD, a utilidade do estudo genético continua por estabelecer em definitivo. A implicação prognóstica da identificação precoce de indivíduos geneticamente afetados é pouco clara dada a baixa penetrância e a elevada expressividade variável relacionada com a idade. Uma minoria imprevisível de doentes sofre eventos arrítmicos na ausência de sintomas prévios ou de evidência clínica da doença. Por outro lado, muitos não desenvolvem doença clinicamente significativa e a maioria dos que a desenvolvem tem um curso relativamente benigno6,10. Neste contexto a sua realização foi principalmente motivada devido à história familiar, não tendo o seu resultado interferido com a abordagem terapêutica adotada.
Relativamente à abordagem terapêutica é discutível a utilização de antiarrítmico numa doente assintomática com atividade ectópica ventricular. De acordo com as guidelines, a utilização de amiodarona ou sotalol tem indicação apenas para o tratamento de TV ou de fibrilhação ventricular quando a implantação de um CDI não é exequível – classe de recomendação IIa, nível de evidência C1. Este facto foi levado em conta na revisão crítica interna à abordagem da situação. A implantação de CDI após a constatação de TV ou de fibrilhação ventricular (FV) − prevenção secundária – tem classe de recomendação I e nível de evidência B; no contexto de prevenção primária e aquando da existência de alterações de alto risco como envolvimento extenso do VD, envolvimento do VE ou síncope inexplicada, mas presumivelmente causada por taquiarritmia tem classe de recomendação IIa com nível de evidência C1.
Este caso clínico permite refletir acerca da importância dos dados fornecidos pela história familiar e dos dados fornecidos pelos meios complementares de diagnóstico que se encontram amplamente disponíveis como o eletrocardiograma e o ecocardiograma, ainda que o primeiro possa ser realizado com recurso a diferentes técnicas, de execução simples e que podem fornecer informação relevante para o diagnóstico desta miocardiopatia mediante a demonstração de um critério major para o seu diagnóstico; por conseguinte, um dos focos de principal interesse deste artigo reside no potencial contributo do ECG modificado de Fontaine no diagnóstico da MAVD. Outro dos pontos de interesse reside na história natural da doença tendo‐se verificado a manifestação de taquidisritmia ventricular potencialmente fatal que é também é critério major para o seu diagnóstico.
Responsabilidades éticasProteção de pessoas e animaisOs autores declaram que para esta investigação não se realizaram experiências em seres humanos e/ou animais.
Confidencialidade dos dadosOs autores declaram que não aparecem dados de pacientes neste artigo.
Direito à privacidade e consentimento escritoOs autores declaram ter recebido consentimento escrito dos pacientes e/ou sujeitos mencionados no artigo. O autor para correspondência deve estar na posse deste documento.
Conflito de interessesOs autores declaram não haver conflito de interesses.
