





Arrhythmogenic right ventricular cardiomyopathy, also known as arrhythmogenic right ventricular dysplasia, is a condition in which myocardium is replaced by fibrous or fibrofatty tissue, predominantly in the right ventricle. It is clinically characterized by potentially lethal ventricular arrhythmias, and is a leading cause of sudden cardiac death. Its prevalence is not known exactly but is estimated at approximately 1:5000 in the adult population. Diagnosis can be on the basis of structural and functional alterations of the right ventricle, electrocardiographic abnormalities (including depolarization and repolarization alterations and ventricular arrhythmias) and family history. Diagnostic criteria facilitate the recognition and interpretation of non-specific clinical features of this disease.
The authors present a case in which the diagnosis of arrhythmogenic right ventricular cardiomyopathy was prompted by the suspicion of right ventricular disease on transthoracic echocardiography. This was confirmed by detection of epsilon waves on analysis of the ECG, which generally go unnoticed but in this case were the key to the diagnosis. Their presence was also shown by non-conventional ECG techniques such as modified Fontaine ECG.
The course of the disease culminated in the occurrence of ventricular tachycardia, which prompted placement of an implantable cardioverter-defibrillator.
A miocardiopatia arritmogénica do ventrículo direito – também denominada de displasia arritmogénica do ventrículo direito – é uma patologia em que se verifica a substituição do miocárdio por tecido fibroso ou fibroadiposo predominantemente no ventrículo direito e que se caracteriza clinicamente por arritmias ventriculares potencialmente letais, sendo uma das causas mais relevantes de morte súbita cardíaca. A sua prevalência exata é desconhecida, no entanto, estima-se que seja de cerca de 1: 5.000 na população adulta. O diagnóstico pode ser efetuado mediante a constatação de alterações estruturais e funcionais do ventrículo direito, alterações eletrocardiográficas – da condução em ECG basal, arritmias ventriculares – e da história familiar. A existência de critérios de diagnóstico facilita o reconhecimento e interpretação das características clínicas não específicas desta entidade.
Apresenta-se um caso clínico em que o diagnóstico de miocardiopatia arritmogénica do ventrículo direito foi desencadeado a partir da suspeita de patologia ventricular direita suscitada por ecocardiograma transtorácico. A análise do ECG serviu para o confirmar, tendo-se detetado ondas épsilon que em geral passam despercebidas, mas que neste caso foram a chave para o diagnóstico tendo-se também aferido a sua existência em técnicas de ECG não convencional como o ECG modificado de Fontaine.
A evolução subsequente do quadro clínico culminou com a ocorrência de taquicardia ventricular o que motivou a implantação de cardiodesfibrilhador implantável (CDI).
A 46-year-old woman was referred for cardiology consultation due to dilatation of the right chambers detected on routine transthoracic echocardiography. The patient was asymptomatic and had no personal history of heart disease; however, she had a daughter diagnosed with arrhythmogenic right ventricular cardiomyopathy (ARVC) with left ventricular involvement, who had an implantable cardioverter-defibrillator (ICD) and was being followed in a different hospital, and another daughter without disease.
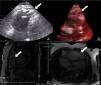
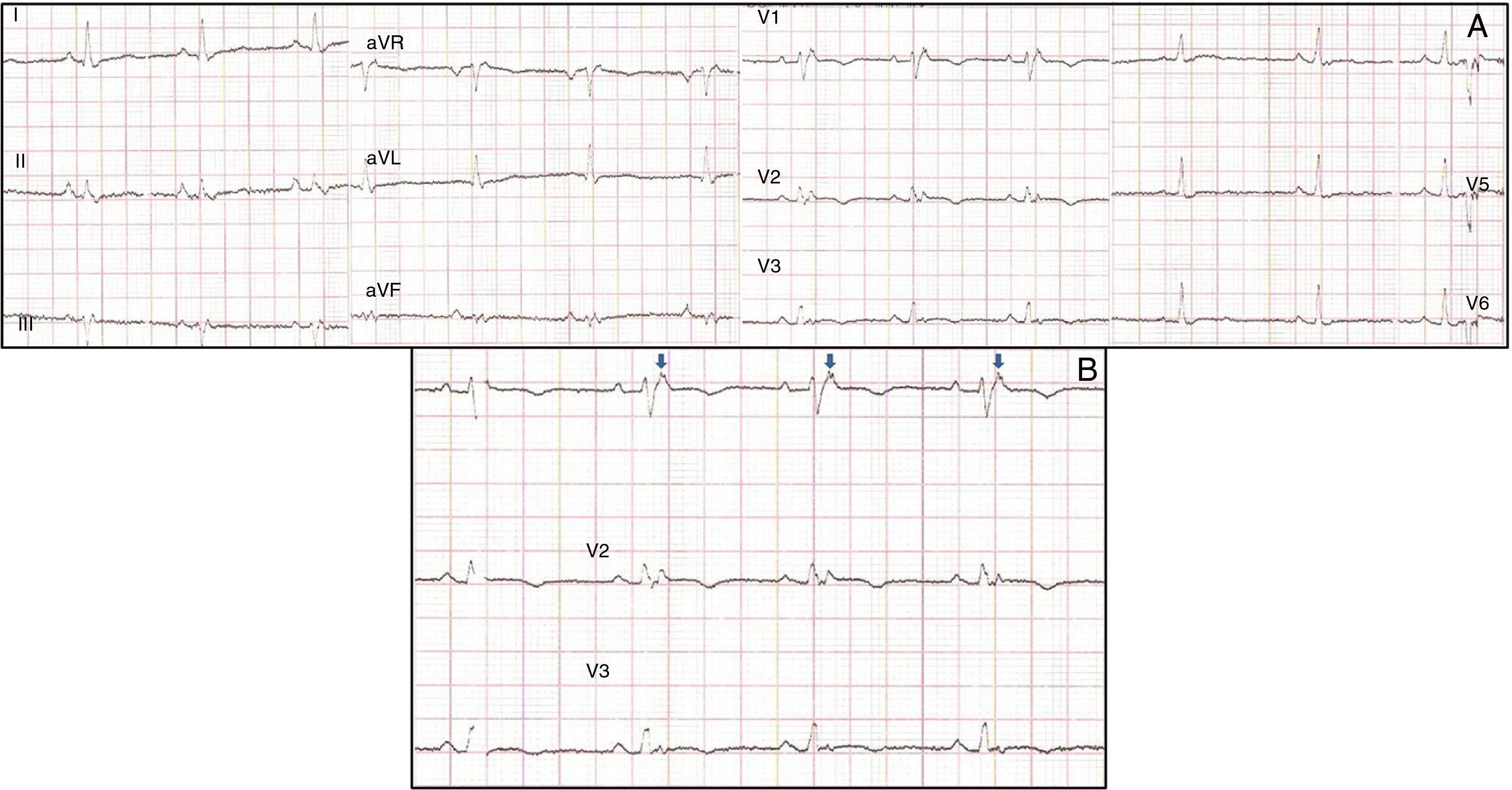
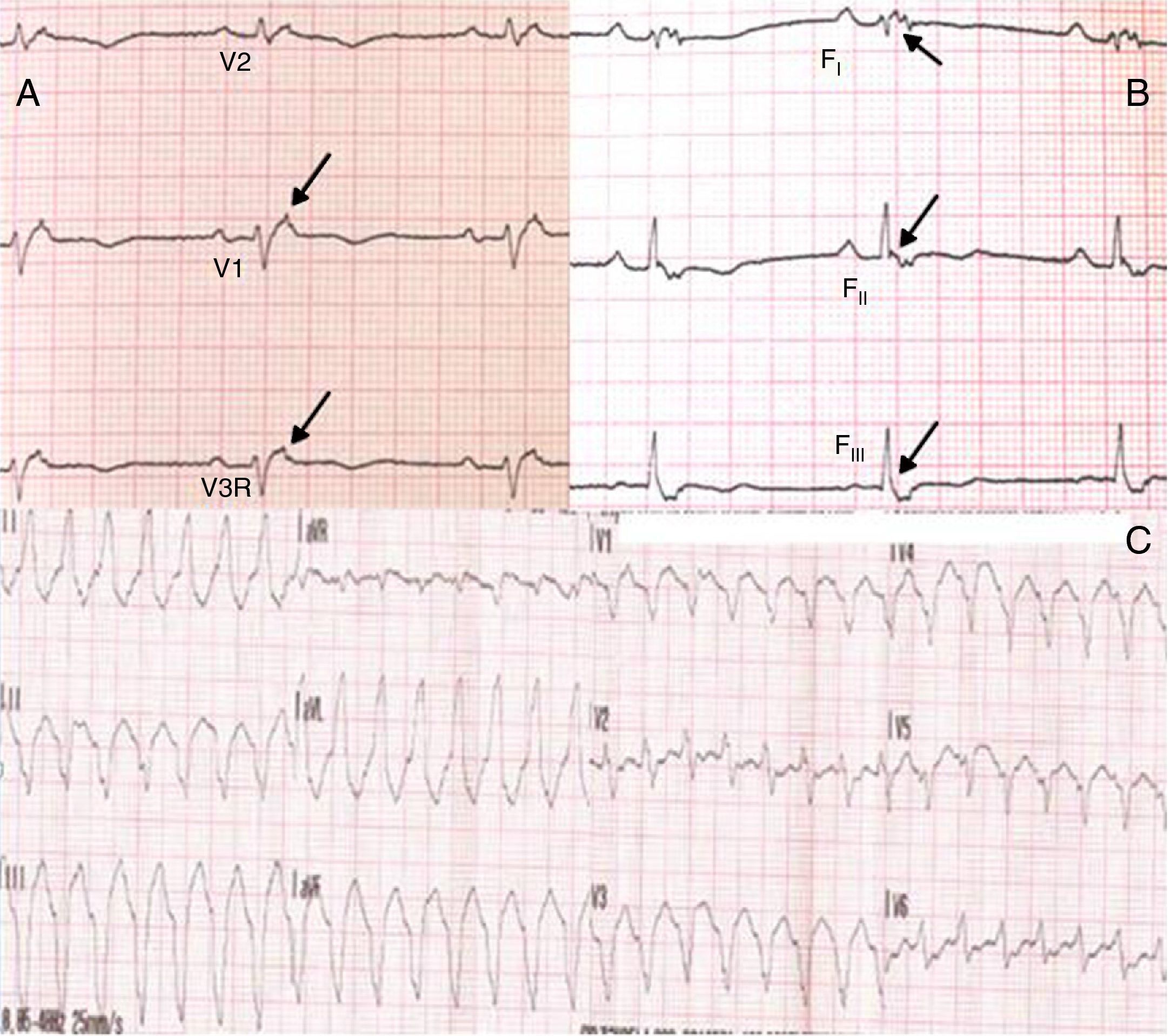
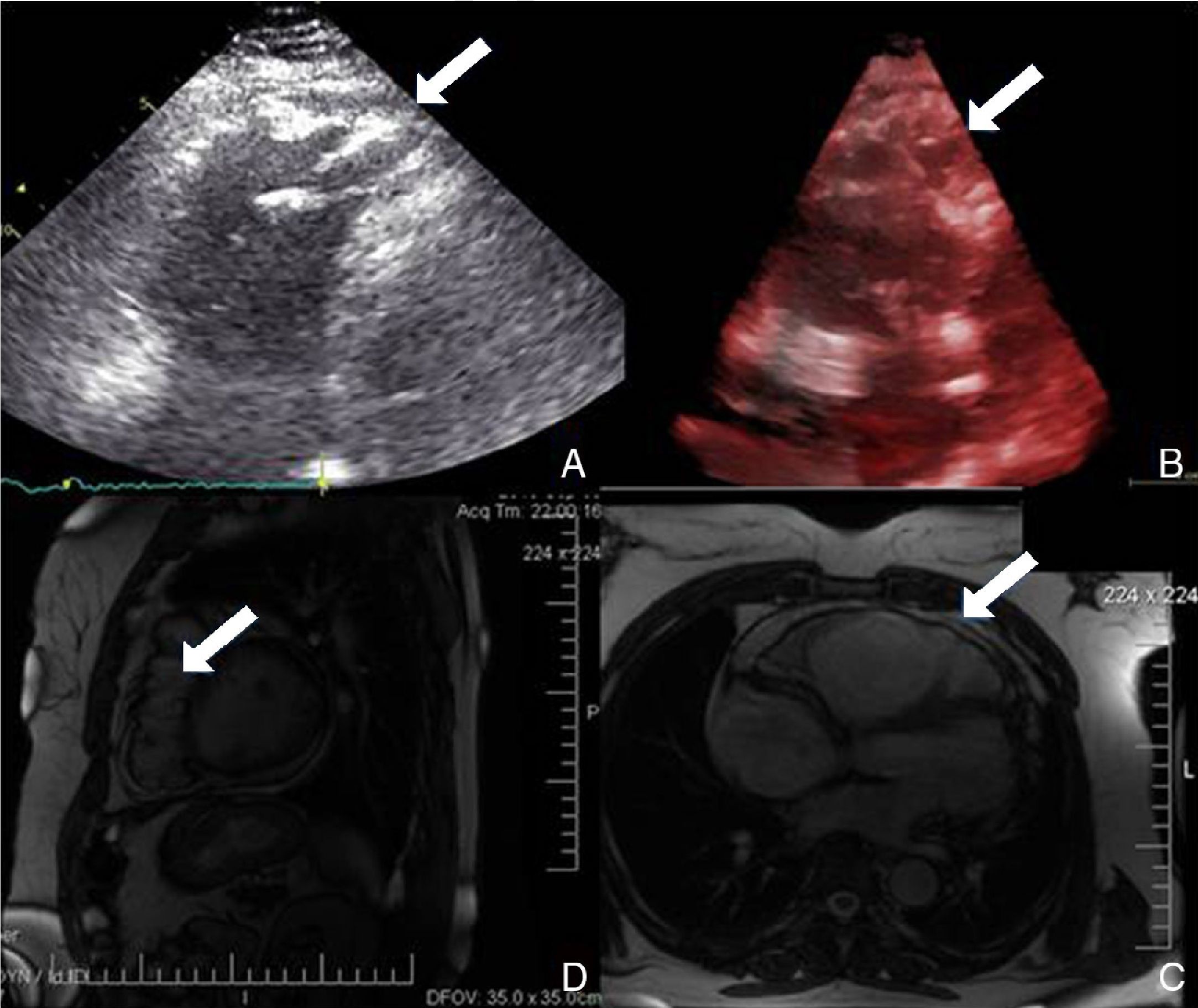
Physical examination showed no abnormalities. Diagnostic exams included 12-lead electrocardiogram (ECG), which showed a pattern of right bundle branch block, together with epsilon waves and T-wave inversion in leads V1–V3 (Figure 1). In order to characterize the typical ARVC alterations, a right-sided precordial lead ECG and a modified Fontaine ECG were performed, the latter with the following placement of electrodes: the right arm electrode over the manubrium, the left arm electrode over the xiphoid and the left leg electrode in the area corresponding to V4, at a recording speed of 25 mm/s and voltage of 10 mm/mV (Figure 2). Both methods clearly showed epsilon waves, especially the modified Fontaine ECG (Figure 3A and B). Two- and three-dimensional transthoracic echocardiography at our institution showed severe right ventricular dilatation and hypokinesia, with prominent apical trabeculae and false tendons and saccular dilatations in the ventricular free wall (Figure 4A and B). Cardiac magnetic resonance imaging revealed small foci of subepicardial fatty infiltration in the right ventricular free and inferior walls, interventricular septum and left ventricular free wall; late enhancement study showed focal enhancement in the interventricular septum consistent with fibrosis (Figure 4C and D).
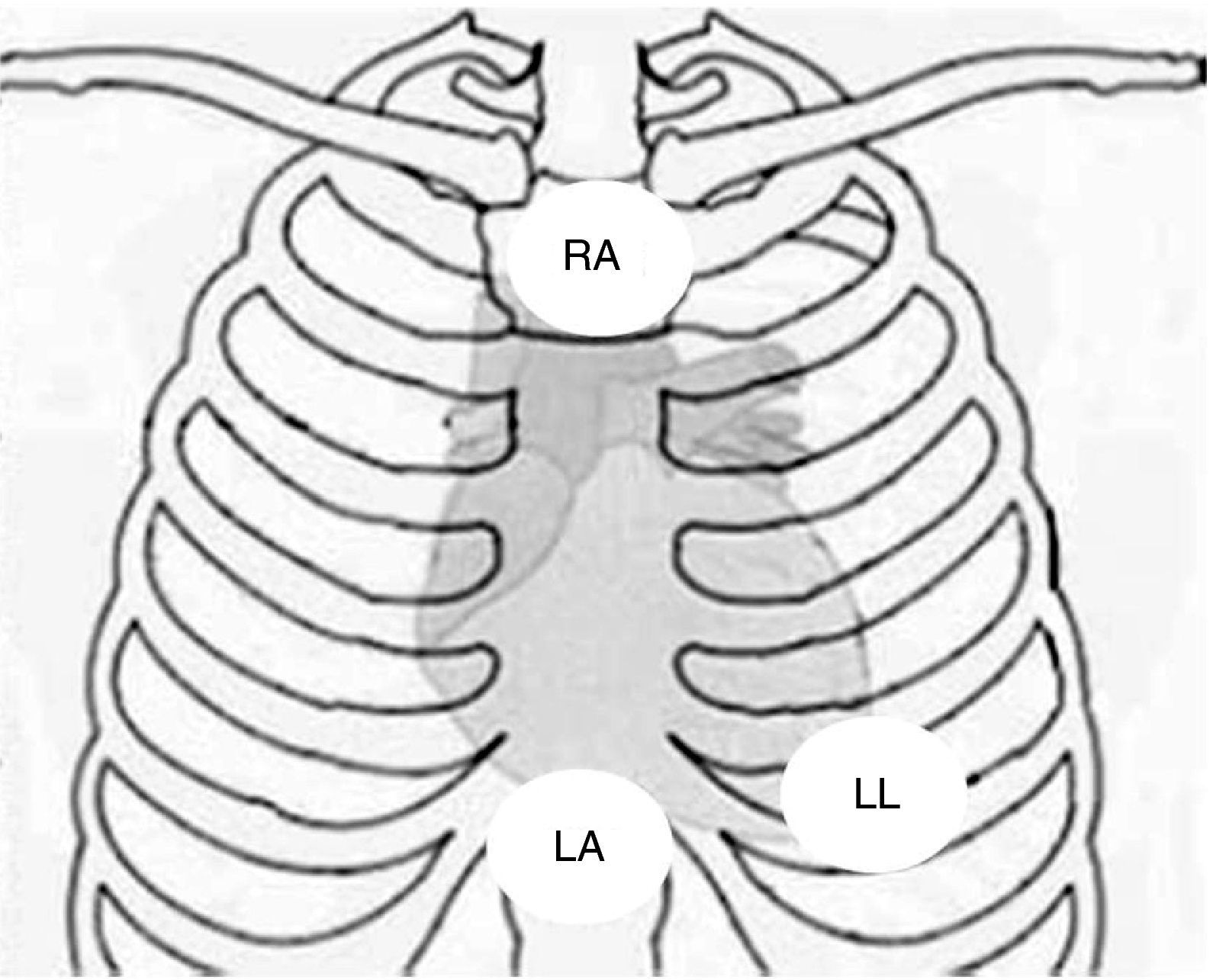
Placement of electrodes in Fontaine ECG. LA: left arm electrode; LL: left leg electrode; RA: right arm electrode.
Screening for mutations in genes coding for desmosomal proteins – plakophilin 2 (PKP2), desmoglein 2 and desmoplakin – revealed a nonsense mutation in exon 12 of the PKP2 gene, also present in the affected daughter. This mutation is found in 11–43% of patients with ARVC.
Since the patient remained asymptomatic and 24-hour Holter monitoring showed only pairs and a triplet of ventricular extrasystoles, therapy with amiodarone 200 mg daily was begun and follow-up was scheduled. Around a year later, following an episode of fainting, nonsustained ventricular tachycardia (VT) was documented, with a pattern of left bundle branch block and superior axis (Figure 3C). This situation, besides being a major criterion for a diagnosis of ARVC, prompted placement of an ICD; around a month after implantation, an episode of VT was converted to sinus rhythm with an appropriate shock following unsuccessful antitachycardia pacing. Around 18 months after this episode, there has been no recurrence of ventricular arrhythmias.
DiscussionARVC is characterized by ventricular arrhythmias and ventricular disease which is reflected macroscopically by replacement of myocardium by fibrous or fibrofatty tissue, predominantly in the right ventricle but sometimes also involving the left ventricle.1,2
Its estimated prevalence is 1:5000 in the general population2 and it is a leading cause of sudden cardiac death (SCD), with an estimated incidence of 0.08–9%.1 However, it may be a contributing factor in up to 10.8% of cases of SCD in young adults.3
Clinical presentation is usually between the ages of 10 and 50, with a mean age at diagnosis of 30.4 The main symptoms are dizziness, palpitations and syncope,4 but most patients are asymptomatic and the diagnosis is suspected following nonspecific ECG alterations, echocardiographic abnormalities or documented ventricular arrhythmias.4
It is estimated that around 30% of cases are familial. Of the two inheritance patterns, autosomal dominant is more common, while in the autosomal recessive form, termed Naxos disease, ARVC is part of a cardiocutaneous syndrome that includes palmoplantar keratoderma and woolly hair.
ARVC should be considered in patients with symptomatic or asymptomatic VT of left bundle branch block morphology in the absence of apparent heart disease.5 A definitive diagnosis requires histological evidence of replacement of right ventricular (RV) myocardium by fibrofatty tissue; however, assessment of this criterion is not practical in clinical practice, and so other data are used for the purpose. The first consensus document detailing diagnostic criteria for this entity appeared in 1994, and a revised version was published in 20106 (Table 1). A definitive diagnosis is made on the basis of two major criteria, one major and two minor criteria, or four minor criteria in different categories. A borderline diagnosis requires one major and one minor criterion or three minor criteria in different categories, and one major criterion or two minor criteria in different categories indicates a possible diagnosis. In the case presented, the patient met the criteria of a family history of ARVC and epsilon waves on 12-lead ECG, which were sufficient for the diagnosis. The latter ECG finding is found in 30% of patients with ARVC, and reflects low-amplitude potentials due to late activation of some parts of the right ventricle. Typically, the epsilon waves are most easily identified in leads V1 to V3, as well as in the right precordial leads, by doubling the sensitivity of the recording and using a filter setting of 40 Hz instead of 150 Hz to decrease the noise level.7 Nevertheless, they are not specific for ARVC since they may be found in cases of RV abnormalities arising from myocardial infarction or cardiac sarcoidosis.7 The usefulness of the Fontaine ECG resides in its greater ability to unmask epsilon waves compared to standard 12-lead ECG and right-sided precordial lead ECG; according to Wang et al., the modified Fontaine ECG doubles or triples the rate of detection of epsilon waves compared to standard 12-lead ECG.8 One explanation for this may be that it records potentials developed in the right ventricle from the infundibulum to the area of the diaphragm.8 This method is thus particularly useful in cases of suspected ARVC, particularly in the presence of right bundle branch block, in which the detection of epsilon waves is even more important since a QRS duration >110 ms does not allow other ECG criteria that are potentially useful in diagnosing this entity to be assessed.7 Although in this particular case the Fontaine ECG was not crucial to the diagnosis, its ability to detect epsilon waves was the main reason for using this simple but little known electrocardiographic technique.
2010 Revised Task Force diagnostic criteria for arrhythmogenic right ventricular cardiomyopathy.
| I. Global and/or regional dysfunction and structural alterations | |
| Major | By 2D echocardiography:- Regional RV akinesia, dyskinesia or aneurysm- and one of the following (end diastole):– PLAX RVOT ≥32 mm (corrected for body size [PLAX/BSA] ≥19 mm/m2)– PSAX RVOT ≥36 mm (corrected for body size [PSAX/BSA] ≥21 mm/m2)– or fractional area change ≤33% |
| By MRI:- Regional RV akinesia, dyskinesia or dyssynchronous RV contraction- and one of the following:– Ratio of RV end-diastolic volume to BSA ≥110 ml/m2 (male) or ≥100 ml/m2 (female)– or RV ejection fraction ≤40% | |
| By RV angiography:- Regional RV akinesia, dyskinesia or aneurysm | |
| Minor | By 2D echocardiography:- Regional RV akinesia or dyskinesia- and one of the following (end diastole):– PLAX RVOT ≥29 to <32 mm (corrected for body size [PLAX/BSA] ≥16 to <19 mm/m2)– PSAX RVOT ≥32 to <36 mm (corrected for body size [PSAX/BSA] ≥18 to <21 mm/m2)– or fractional area change >33% to ≤40% |
| By MRI:- Regional RV akinesia or dyskinesia or dyssynchronous RV contraction- and one of the following:– Ratio of end-diastolic volume to BSA ≥100 to <110 ml/m2 (male) or ≥90 to <100 ml/m2 (female)– or fractional area change >40% to ≤45% | |
| II. Tissue characterization of ventricular wall | |
| Major | Residual myocytes <60% by morphometric analysis (or <50% if estimated), with fibrous replacement of the RV free wall myocardium in ≥1 sample, with or without fatty replacement of tissue on endomyocardial biopsy |
| Minor | Residual myocytes 60% to 75% by morphometric analysis (or 50% to 65% if estimated), with fibrous replacement of the RV free wall myocardium in ≥1 sample, with or without fatty replacement of tissue on endomyocardial biopsy |
| III. Ventricular repolarization abnormalities | |
| Major | - Inverted T waves in right precordial leads (V1, V2 and V3) or beyond in individuals >14 years of age (in the absence of complete right bundle branch block with QRS ≥120 ms). |
| Minor | - Inverted T waves in leads V1 and V2 in individuals >14 years of age (in the absence of complete right bundle branch block with QRS ≥120 ms) or in V4, V5 or V6- Inverted T waves in leads V1, V2, V3 and V4 in individuals >14 years of age in the presence of complete right bundle branch block with QRS ≥120 ms |
| IV. Depolarization/conduction abnormalities | |
| Major | - Epsilon wave in the right precordial leads (V1 to V3) |
| Minor | - Late potentials by SAECG in ≥1 of 3 parameters in the absence of a QRS duration ≥110 ms on the standard 12-lead ECG– Filtered QRS duration (fQRS) ≥114 ms– Duration of terminal QRS <40 μV (low-amplitude signal duration) ≥38 ms– Root-mean-square voltage of terminal 40 ms ≤20 μV– Terminal activation duration of QRS ≥55 ms measured from the nadir of the S wave to the end of the QRS, including R′, in V1, V2, V3, in the absence of complete right bundle branch block. |
| V. Arrhythmias | |
| Major | - Nonsustained or sustained ventricular tachycardia of left bundle-branch morphology with superior axis (negative or indeterminate QRS in leads II, III, and aVF and positive in lead aVL) |
| Minor | - Nonsustained or sustained ventricular tachycardia of RV outflow configuration, left bundle-branch block morphology with inferior axis (positive QRS in leads II, III, and aVF and negative in lead aVL) or of unknown axis- >500 ventricular extrasystoles per 24 hours (Holter) |
| VI. Family history | |
| Major | - ARVC confirmed in a first-degree relative who meets Task Force criteria- ARVC confirmed pathologically at autopsy or surgery in a first-degree relative- Identification of a pathogenic mutationa categorized as associated or probably associated with ARVC in the patient under evaluation |
| Minor | - History of ARVC in a first-degree relative in whom it is not possible or practical to determine whether the family member meets Task Force criteria- Premature sudden death (<35 years of age) due to suspected ARVC in a first-degree relative- ARVC confirmed pathologically or by current Task Force criteria in second-degree relative |
ARVC: arrhythmogenic right ventricular cardiomyopathy; BSA: body surface area; MRI: magnetic resonance imaging; PLAX: parasternal long-axis view; PSAX: parasternal short-axis view; RV: right ventricular; RVOT: right ventricular outflow tract; SAECG: signal-averaged ECG.
A pathogenic mutation is a DNA alteration associated with ARVC that alters or is expected to alter the encoded protein, is unobserved or rare in a large non-ARVC control population, and either alters or is predicted to alter the structure or function of the protein or has demonstrated linkage to the disease phenotype in a conclusive pedigree.
While magnetic resonance imaging was not mandatory in the case presented following transthoracic echocardiography, we decided to perform it due to its feasibility and its value in characterization of the imaging features of this cardiomyopathy.9
Although mutations in the genes coding for desmosomal proteins are relatively common in patients with suspected ARVC, the usefulness of genetic study remains the subject of debate. The prognostic implications of early identification of affected individuals are unclear given its low penetrance and highly variable expression according to age. A minority of patients suffer arrhythmic events in the absence of previous symptoms or clinical signs of the disease. Many do not in fact develop clinically significant disease, and most of those that do have a relatively benign course.6,10 In the present case, genetic study was performed mainly due to the patient's family history, and its results did not affect the therapeutic approach adopted.
With regard to therapeutic management, it is debatable whether antiarrhythmics should be used in asymptomatic patients with ventricular ectopic activity. According to the ACC/AHA/ESC guidelines, amiodarone and sotalol are only indicated (class IIa recommendation, level of evidence C) to treat VT or ventricular fibrillation (VF) when an ICD is not possible,1 a fact that was taken into account during internal review of the approach to adopt in the present case. ICD implantation for secondary prevention following documented VT or VF is a class I recommendation, level of evidence B, while implantation for primary prevention in patients with high-risk alterations such as extensive RV involvement, left ventricular involvement or unexplained syncope assumed to be due to tachyarrhythmia is a class IIa recommendation, level of evidence C.1
The case presented highlights the importance of information on family history and of the data provided by widely available diagnostic exams such as ECG and echocardiography. Different ECG techniques that are simple to perform can be useful for initial assessment by demonstrating the presence of a major diagnostic criterion for this cardiomyopathy. Thus, one of the main points of interest in this case report is the potential role of modified Fontaine ECG leads in the diagnosis of ARVC; another is the natural history of the disease, with the patient suffering potentially fatal VT, a major criterion for the diagnosis.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that no patient data appear in this article.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Moreira D, Delgado A, Marmelo B, et al. Miocardiopatia arritmogénica do ventrículo direito. Contribuição de diferentes técnicas de eletrocardiografia. Rev Port Cardiol. 2014;33:243.e1–243.e7.
