




O recetor da angiotensina do tipo 2, AT2R, tem vindo a ser descrito como tendo ações opostas ao recetor da angiotensina do tipo 1, AT1R. Apesar do AT2R existir em baixas quantidades no adulto, a sua expressão sobe bastante em situações patológicas. O AT2R tem três grandes vias de sinalização: a ativação fosfátases de serina/treonina (promoção da apoptose celular e efeitos antioxidantes); ativação da via bradiquinina/NO/cGMP (promoção de vasodilatação) e ativação da fosfolípase A2 (associada ao controlo das correntes de potássio). O AT2R parece ter um efeito na remodelação vascular, na prevenção da aterosclerose e na descida da pressão sanguínea (quando associada a um inibidor do AT1R). Após enfarte do miocárdio, o AT2R parece diminuir o tamanho do enfarte, a hipertrofia cardíaca, a fibrose e aumentar a função cardíaca. Contudo, o seu papel a nível cardíaco é o mais controverso. A nível renal o AT2R promove a natriurese. Até agora, a terapêutica direcionada para o sistema renina‐angiotensina‐aldosterona é à base de inibidores da enzima de conversão da angiotensina (IECA) ou de inibidores do recetor da angiotensina tipo 1 (ARA). O estudo do AT2R foi revolucionado pela descoberta de um agonista direto, o C21, que promete integrar parte da terapêutica das doenças cardiovasculares.
The angiotensin type 2 receptor, AT2R, has been described as having opposite effects to the angiotensin type 1 receptor, AT1R. Although the quantities of the AT2R found in the adult are low, its expression rises in pathological situations. The AT2R has three major signaling pathways: activation of serine/threonine phosphatases (promoting apoptosis and antioxidant effects), activation of the bradykinin/NO/cGMP pathway (promoting vasodilation), and activation of phospholipase A2 (associated with regulation of potassium currents). The AT2R appears to have effects in vascular remodeling, atherosclerosis prevention and blood pressure lowering (when associated with an AT1R inhibitor). After myocardial infarction, the AT2R appears to decrease infarct size, cardiac hypertrophy and fibrosis, and to improve cardiac function. However, its role in the heart is controversial. In the kidney, the AT2R promotes natriuresis. Until now, treatment directed at the renin‐angiotensin‐aldosterone system has been based on angiotensin‐converting enzyme inhibitors or angiotensin type 1 receptor blockers. The study of the AT2R has been revolutionized by the discovery of a direct agonist, C21, which promises to become part of the treatment of cardiovascular disease.
No âmbito do sistema renina‐angiotensina‐aldosterona (SRAA), o recetor da angiotensina (Ang) II mais amplamente estudado é o AT1R. Este recetor é o responsável pela maioria das consequências da ativação deste sistema tais como: vasoconstrição, retenção de sódio, estimulação da libertação de aldosterona, proliferação celular, hipertrofia cardíaca e vascular, participação no stress oxidativo e inflamação. A Ang II pode‐se ligar a um segundo recetor, o AT2R, cuja existência já se descobriu há muito tempo1. Contudo, as funções ainda não se apresentam completamente clarificadas. A ativação deste recetor tem sido descrita como tendo uma ação contrária à da ativação do AT1R2–5. Assim, o AT2R parece desempenhar uma função protetora em patologias como a hipertensão, a aterosclerose e o enfarte do miocárdio (EM). O AT1R e o AT2R são ambos recetores transmembranares acoplados à proteína G (G protein coupling receptors [GPCR])6 e possuem uma homologia de sequência de 34%7. Com estes recetores também interagem várias GPCR interacting proteins (GIP) que influenciam a sua atividade, ligando‐se ao terminal carboxilo8,9. No adulto o AT1R é expresso ubiquamente, enquanto o AT2R existe em baixas quantidades, mas principalmente nos vasos sanguíneos, nos rins, na medula da glândula suprarrenal, no útero, nos ovários, no coração e em núcleos especializados no cérebro2,10. Todavia, a expressão de AT2R aumenta nas situações patológicas já referidas. Assim, o papel fisiológico do AT2R não é significativo. No feto, verifica‐se o oposto, ou seja, uma maior prevalência de AT2R, que poderá estar relacionado com a sua eventual função no desenvolvimento fisiológico geral. Esta revisão tem por objetivo abordar as funções do AT2R no sistema cardiovascular, relacionando‐as com possíveis vias terapêuticas.
Vias de transdução de sinal do recetor tipo 2 da angiontensinaOs recetores da Ang II formam homodímeros e heterodímeros entre si. O homodímero AT1R/AT1R e o homodímero AT2R/ AT2R promovem os efeitos dos recetores respetivos9. Já quando há formação do heterodímero AT1R/AT2R, a sinalização do AT1R está diminuída, sendo esta uma forma pela qual o AT2R inibe diretamente os efeitos do AT1R11. Entretanto, também pode haver heterodimerização do recetor AT2R com o recetor da bradiquinina tipo 2 (B2R), promovendo a produção de óxido nítrico12. Esta interação revela‐se importante no sistema quinina/NO/cGMP.
Uma vez ativo, o AT2R tem, essencialmente, três vias de sinalização: ativação de fosfátases de serina/treonina, ativação da via bradiquinina/NO/cGMP e ativação da fosfolípase A213 (ver Figura 1).
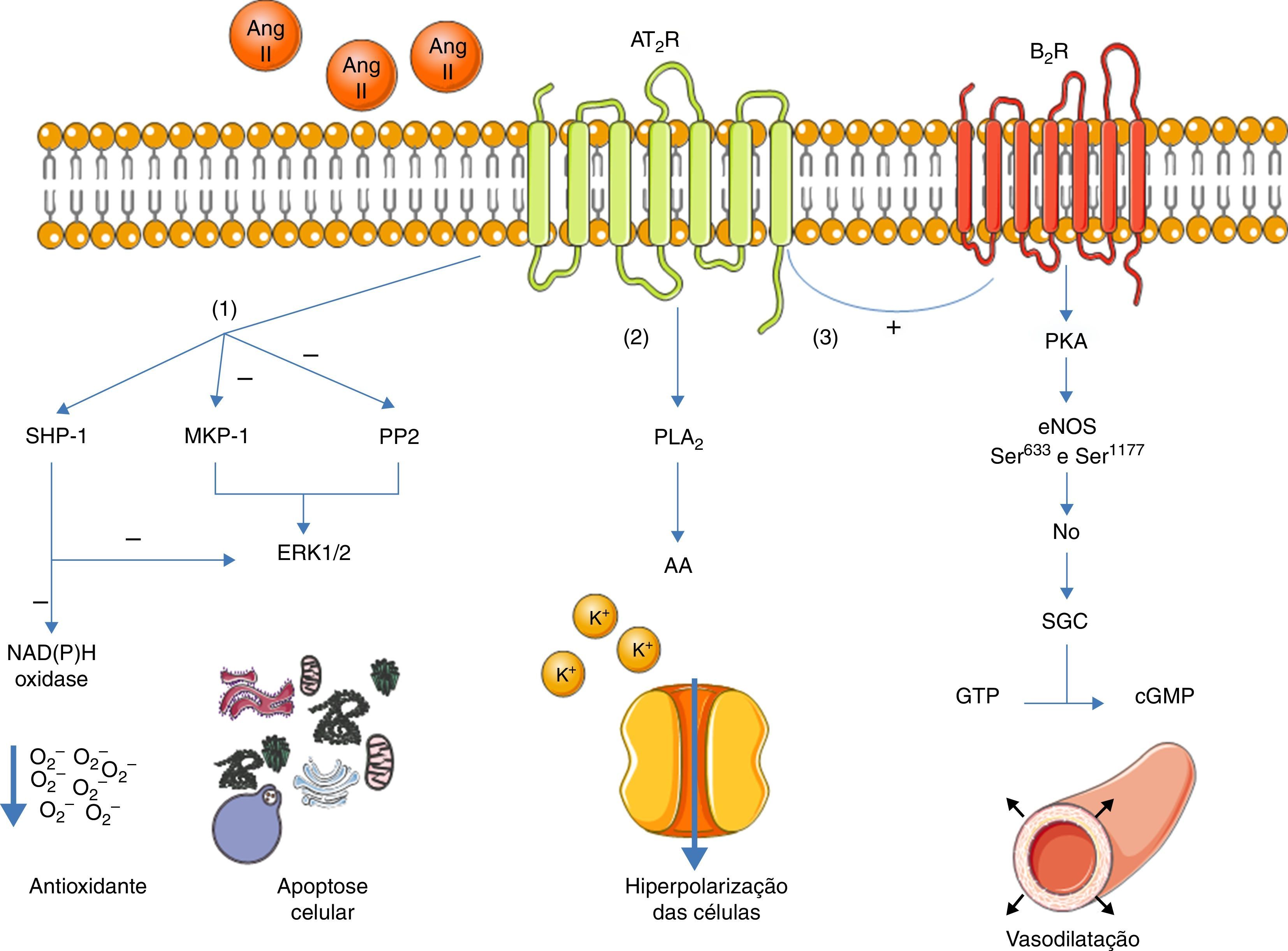
Principais vias de sinalização do AT2R. A ativação do AT2R leva a três principais vias de sinalização: ativação de fosfátases de serina/treonina (1); ativação da fosfolípase A2(2) e ativação da via bradiquinina/NO/cGMP (3).
Legenda: AA: arachidonic acid; Ang II: angeotensinII; AT2R: angiotensin receptor type 2; B2R: bradykinin receptor B2; cGMP: cyclic guanosine monophosphate; eNOS: endothelial nitric oxide synthase; ERK1/2: extracellular‐regulated kinase 1 and 2; GTP: guanosine triphosphate; MKP‐1: MAP kinase phosphatase; NO: nitric oxide; PLA2: phospholipase A2; PKA: protein kinase A; PP2: protein phosphatase 2; sGC: soluble guanylate cyclase; SHP‐1: SH2 domain containing tyrosine phosphatase. Os componentes das ilustrações são retirados do website: http://www.servier.co.uk/medical‐art‐gallery/.
No que concerne às fosfátases, foi estudado o papel do AT2R na ativação da MAP kinase phosphatase (MKP‐1), da protein phosphatase 2 (PP2A) e da SH2 domain containing tyrosine phosphatase (SHP‐1)3,13.
A ativação da MKP‐1 e da PP2A pelo AT2R resulta na inibição das ERK1/2, promovendo a apoptose celular. A ativação da SHP‐1 é responsável pela inibição da ERK1/2 e também inibe a atividade da NAD(P)H oxidase (que é estimulada pelo AT1R14), participando assim na defesa antioxidativa do endotélio15.
A ativação do sistema bradiquinina/NO/cGMP está associada à vasodilatação4,16. O AT2R, quando ativado, estimula o recetor B2R que, por sua vez, estimula a fosforilação da síntase de NO (eNOS) através da PKA (sendo que esta fosforilação ocorre nos resíduos Ser633 e Ser1177)17.
Assim, há aumento da produção de NO, que ativa a guanil ciclase (sGC), que converte o GTP em cGMP. Este último promove a vasodilatação. Apesar de este mecanismo já ter sido descrito16,18, o papel do AT2R no controlo da pressão sanguínea (PS) ainda permanece controverso1.
O AT2R também promove a ativação da fosfolípase A2 (PLA2) e a libertação subsequente de ácido araquidónico (AA). Este AA é responsável pelo controlo das correntes de potássio, levando a uma hiperpolarização e diminuição da excitabilidade das células3,19. Esta ação parece ser particularmente determinante na diminuição da atividade simpática20.
Guilluy et al.21 descreveram ainda uma via de sinalização alternativa ao sistema bradiquinina/NO/cGMP que leva a vasodilatação e ocorre diretamente nas células musculares lisas vasculares (ver Figura 2). Esta via culmina na fosforilação e inativação da RhoA no resíduo Ser188, levando à vasodilatação. Esta fosforilação é feita pela Ste20‐related kinase (SLK) e é independente da eNOS, PKA e PKG. Esta atividade da SLK diminui quando se encontra fosforilada. Ou seja, é no estado desfosforilado que a SLK fosforila a RhoA. Entretanto, a cínase da caseína 2 (CK2) é responsável pela fosforilação basal da SLK. Sendo assim, o papel do AT2R nesta via é diminuir a atividade da CK2 de forma a aumentar a quantidade de SLK desfosforilada. Como a CK2 se encontra ativa quando fosforilada, o AT2R consegue o efeito inibitório através do SHP‐1 que desfosforila a CK2.
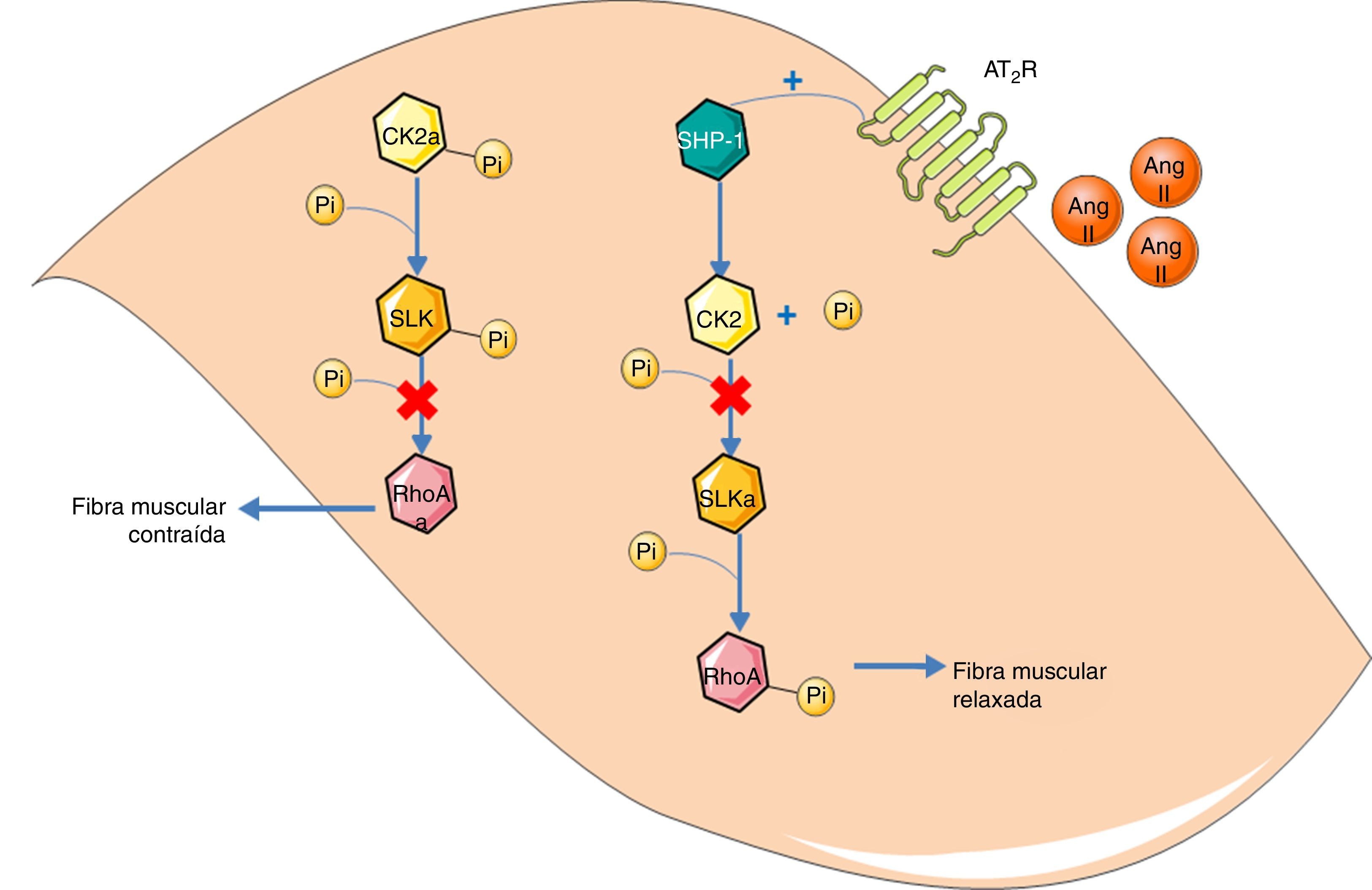
Via alternativa de vasodilatação mediada pelo AT2R. A ativação do AT2R leva à ativação SHP‐1. Esta, por sua vez, desfosforila a CK2, inativando‐a. A CK2, já não fosforila, e inativa, a SLK. A SLKa fosforila a RhoA, inativando‐a, promovendo assim relaxamento da fibra muscular lisa vascular. O sufixo «a» significa «ativada».
Legenda: Ang II: angeotensin II; AT2R: angiotensin receptor type 2; CK2: casein kinase 2; Pi: inorganic phosphate; RhoA: Ras homolog family member A; SHP‐1: SH2 domain containing tyrosine phosphatase; SLK: Ste20‐related kinase. Os componentes das ilustrações são retirados do website: http://www.servier.co.uk/medical‐art‐gallery/.
O AT2R participa também em vias anti‐inflamatórias que levam à diminuição da expressão da interleucina‐6 (IL‐6)22. O AT2R participa essencialmente na estimulação de fosfátases e serina/treonina e na síntese de AA que, de seguida, dá origem ao metabolito 11,12‐EET, que vai participar na via inflamatória22. Estes dois processos convergem na redução da atividade do fator nuclear kB (NF‐kB), o que diminui a IL‐622.
É de referir que também ocorre uma ativação destes recetores, independentemente da ligação de Ang II. O AT1R pode ser ativado por stress mecânico, sem que haja a ligação da Ang II23. Esta ativação leva ao aumento da expressão de calcineurina que, por sua vez, é responsável pela hipertrofia cardíaca23. Por outro lado, Miura e Karnik24 demonstraram, num estudo em ovários de rato, que o AT2R não necessita da Ang para induzir a apoptose. Atualmente é aceite que parte do AT2R se pode apresentar constitutivamente ativo na forma de homo‐oligómeros25. Estas particularidades destes recetores da Ang II são especialmente relevantes na sua abordagem farmacológica, como também será aqui discutido.
Recetor tipo 2 da angiontensina e o agonista C21Os estudos passados sobre o AT2R foram sempre feitos de forma indireta. Alguns estudos ativavam seletivamente o AT2R, fornecendo Ang II na presença de um inibidor do AT1R. Outra alternativa era um antagonista do AT2R como o PD123319 e estudar os efeitos da ausência de função deste recetor. Todavia, este PD123319 tem ainda o problema de ter falta de especificidade quando aplicado em altas doses1. Outros estudos modificavam ratinhos geneticamente para não expressarem ou para sobre‐expressarem o gene do AT2R.
A abordagem mais direta para estudar o AT2R é a utilização de um agonista específico do AT2R. Porém, até 2004, o melhor agonista que existia era o CGP42112A. No entanto, este ligando degrada‐se rapidamente in vivo e para além de agonista também tem propriedades antagónicas1. Foi então que surgiu um novo agonista que está a revolucionar, desde então, o estudo do AT2R: o compound 21 (C21) – Wan et al.26. Este é um agonista não‐peptídico, que pode ser administrado por via oral e permite a estimulação do AT2R in vivo e in vitro, sem afetar o AT1R.
Efeitos vasculares do recetor tipo 2 da angiontensinaA Ang II regula vários processos implicados na fisiopatologia vascular, tais como: crescimento/apoptose das células vasculares, migração das células musculares lisas vasculares, respostas inflamatórias e remodelação da matriz extracelular13. Estes efeitos são contrabalançados pela ligação da Ang II ao AT1R ou ao AT2R, que têm ações opostas. No que toca à regulação da PS, uma deleção do gene do AT2R está associada a um aumento da PS4,27, isto, em virtude da vasodilatação promovida por este recetor. Um estudo que marcou este tema foi o de Savoia et al.28, no qual foi demonstrado, pela primeira vez, o aumento da AT2R em artérias periféricas de pacientes diabéticos hipertensos tratados com valsartan (um inibidor do AT1R) durante um ano. Este aumento de AT2R cursou com uma diminuição da PS nestes doentes, dependendo esta descida da quantidade de AT2R expressa. Para confirmar esta ação do AT2R, o grupo recolheu ainda amostras de artérias periféricas pré‐contraídas e expô‐las, ex vivo, em valsartan. O resultado foi, novamente, um aumento da expressão de AT2R e vasodilatação. Verificou‐se ainda, que o aumento do AT2R se deu principalmente na túnica média, tendo os autores avançado que o AT2R pode estimular a vasodilatação diretamente nas células musculares lisas vasculares (via RhoA, já referida anteriormente).
Zhang et al.29 estudaram as funções do AT1R e do AT2R na circulação coronária. O estudo conclui que a estimulação do AT1R provoca vasoconstrição e a estimulação do AT2R provoca vasodilatação. Quando o endotélio foi retirado ou foi administrado um inibidor da síntase de NO, a resposta vasodilatadora do AT2R foi abolida. Os autores também descobriram que a produção de superóxido pela estimulação do AT1R (via estimulação da NAD(P)H oxidase, como já foi referido) leva a uma inibição da vasodilatação. Isto acontece porque o superóxido reage com o NO, funcionando como um «raptor» de NO, impedindo assim a vasodilatação.
O AT2R está presente em grande quantidade na vida fetal e por isso se defende que terá uma função no correto desenvolvimento e crescimento vascular3. De fato, a expressão de AT2R está associada à angiogénese fetal e influencia o fenótipo das células musculares lisas vasculares, por via do controlo das ERK30. A maior expressão de AT2R dá‐se no final do período gestacional e está associada a uma diminuição da síntese de DNA na aorta31. A correta diferenciação vascular é, então, conseguida pelas ações contrárias do AT1R e do AT2R32. Já no adulto, o AT2R continua a participar em processos de remodelação vascular, promovendo a apoptose celular e inibindo a proliferação celular (potenciada pelo AT1R). Isto é particularmente evidente após a ballon‐injury da artéria carótida, sendo que a adição de gene do AT2R in vivo reduz significativamente a formação de neoíntima31,33. O descontrolo desta remodelação está associado a patologias. Por exemplo, a perda do AT2R acelera o desenvolvimento e a rotura de um aneurisma na aorta, em ratinhos com síndrome de Marfan34. Nestes ratinhos, um inibidor do AT1R (ou seja, um ARA, neste caso o losartan) diminuiu a progressão do aneurisma, porém a proteção máxima necessita que o AT2R seja expresso34. Também há indicação que o AT2R participa na proteção contra a hipertrofia vascular35.
Com a descoberta do C21, novos estudos têm surgido e têm‐se obtido novos resultados no âmbito dos efeitos vasculares do AT2R. A ideia que a vasodilatação promovida pelo AT2R está associada a uma redução da PS tem vindo a ser desmentida por estes novos estudos. A administração do C21, por si só, não diminuiu a PS em animais normotensos36,37, nem em animais hipertensos36,38,39. Apenas no estudo que descreveu pela primeira vez o C21 se verificou diminuição da PS em ratos hipertensos26. Porém, este estudo foi feito em ratos anestesiados, o que os torna muito mais sensíveis a interferências farmacológicas no sistema renina angiotensina aldosterona1. Apesar de tudo, é difícil observar os efeitos de uma estimulação direta de AT2R in vivo, dada a predominância do AT1R. Assim, quando o C21 foi administrado juntamente uma baixa dose de um ARA (candesartan), que não modifica a PS por si só, verificou‐se uma queda significativa da PS em ratos hipertensos36. Esta resposta foi extinta quando se administrou um antagonista do AT2R36. Já quando se associou o C21 a uma alta dose de um ARA, não se verificou efeito adicional do C21 na PS, relativamente ao ARA sozinho38,39.
Estes estudos com C21 vieram confirmar que o AT2R atua essencialmente como protetor da lesão vascular. Rehman et al.38 mostraram que a o C21, por si só, promove a diminuição da rigidez vascular em ratos hipertensos. Também provaram que o C21, por si só, leva a menor deposição de colagénio na túnica média da aorta e a menor ratio colagénio perivascular coronário: colagénio da túnica média coronária (em termos de área de seção). Já a experiência de Paulis et al.39 estudou os efeitos do C21, independentes do NO, uma vez que se usou um inibidor da síntase de NO (o L‐NAME) comum a todos os grupos (exceto o grupo de controlo). Concluiu‐se que a estimulação crónica do AT2R pelo C21 previne o aumento da rigidez aórtica e o aumento da deposição de colagénio sem interferir com a hipertensão.
Efeitos do recetor tipo 2 da angiotensina na estrutura cardíacaTambém no coração os dois recetores da Ang II participam em processos de remodelação. Porém, no caso do coração, ainda não há consenso sobre como estas alterações se processam. Sabe‐se que os AT2R, em condições normais, se expressam no coração em quantidades muito residuais. Porém, aquando de patologia (como hipertensão ou EM), a sua expressão aumenta muito e é nestes casos que a sua ação ganha preponderância. A maior controvérsia acerca das ações do AT2R está em estudos sobre hipertrofia cardíaca, fibrose e função cardíaca no pós‐enfarte1. A remodelação cardíaca após um EM é caracterizada por uma fase aguda de expansão do enfarte, seguida de hipertrofia e fibrose intersticial nos cardiomiócitos que não sofreram isquemia40. A maioria dos estudos apontam para um efeito benéfico AT2R a nível cardíaco. A revisão de Johren et al.3, que data de 2004 (antes da descoberta do C21), fez uma síntese dos resultados de estudos em modelos knockout para o AT2R a nível do miocárdio. Os principais resultados foram: aumento da expressão de AT1R (e, consequentemente, aumento dos seus efeitos proliferativos); aumento da hipertrofia após EM; aumento da rotura cardíaca após EM; redução da sobrevivência e dilatação ventricular esquerda após EM. De entre os principais desacordos, temos estudos que apontam que este knockout para o AT2R promove a fibrose perivascular e outros que concluem exatamente o contrário.
Após esta revisão, novos estudos foram surgindo, mas as controvérsias ainda não se dissiparam. A Tabela 1 apresenta uma compilação dos resultados obtidos em estudos mais recentes. É de destacar o estudo de Voros et al.41 no qual houve comparação de cinco grupos de ratinhos (ver Tabela 1). Quer o bloqueio do AT1R, quer a sobre‐expressão do AT2R, mostraram contribuir com efeitos benéficos para a remodelação cardíaca. Consistentemente, o grupo de ratinhos transgénicos com sobre‐expressão de AT2R + knockouts para o AT1R foi o que mostrou a maior fração de ejeção, o menor volume telediastólico no ventrículo esquerdo e menor fibrose em áreas adjacentes ao enfarte. Surgiram também estudos que contrariam a ideia que o AT2R tem efeitos protetores cardíacos. No estudo de Tschöpe et al.42, após EM, ratinhos knockout para o AT2R não mostraram alterações na função sistólica e diastólica do ventrículo esquerdo, nem alterações na fibrose e na matriz extracelular. Concluíram então que, após EM, a hipertrofia e a fibrose induzidas pela isquemia são independentes do AT2R. Já no estudo de van Esch et al.43, a ausência de AT2R não potenciou os efeitos do AT1R e, na ausência de AT1R, não se verificaram os efeitos cardíacos mediados pelo AT2R.
Efeitos do AT2R após enfarte do miocárdio (EM) e na insuficiência cardíaca (IC)
| Efeitos benéficos após EM (em comparação com o controlo) |
| Diminuição do tamanho do enfarte44,70 |
| Aumento do dP/dt máx e do dP/dt min44,70,71 |
| Aumento da fração de ejeção41,70–73 |
| Aumento da contratibilidade44 |
| Diminuição do volume telessistólico do ventrículo esquerdo41,72,73 |
| Diminuição do volume telediastólico do ventrículo esquerdo41,72,73 |
| Diminuição da pressão telediastólica do ventrículo esquerdo44,71 |
| Diminuição da pressão na aurícula esquerda70* |
| Inibição do crescimento do ventrículo esquerdo74 |
| Diminuição do ratio peso do ventrículo esquerdo/peso corporal74 |
| Diminuição do ratio peso do pulmão/peso corporal74 |
| Dimuição da fibrose intersticial em zonas circundantes do enfarte41 |
| Diminuição dos níveis da monocyte chemoattractant protein‐1, da myeloperoxidase e das interleucinas (efeitos anti‐inflamatórios)44 |
| Ausência de efeitos/efeitos deletérios após EM (em comparação com o controlo) |
| Ausência de alterações na função sistólica e diastólica42 |
| Ausência de alterações na fibrose e na matriz extracelular42 |
| Aumento do volume telessistólico e telediastólico do ventrículo esquerdo45 |
| Ausência de inibição dos efeitos do AT1R43 |
| Efeitos benéficos na IC (em comparação com o controlo)* |
| Aumento do dP/dt máx e do dP/dt min75 |
| Aumento da fração e ejeção75 |
| Diminuição da pressão pulmonar média75 |
| Diminuição do volume telessistólico do ventrículo esquerdo75 |
| Diminuição do volume telediastólico do ventrículo esquerdo75 |
| Diminuição da pressão telediastólica do ventrículo esquerdo75 |
| Diminuição dos aldeídos (marcadores de stress oxidativo)75 |
| Abordagem realizada |
| Ratinhos knockout para o AT2R42 |
| Ratinhos transgénicos com sobre‐expressão de AT2R versus controlo72,73 |
| Ratinhos wilde‐type+ARA versus ratinhos knockout para o AT2R+ARA74 |
| Ratinhos knockout para o AT1R+ wilde‐type para o AT2R versus ratinhos knockout para o AT2R+ wilde‐type para o AT1R43 |
| Ratinhos injetados no ventrículo esquerdo com adeno‐vírus promotor do AT2R71 |
| Bloqueiro do AT1R com ARA em cães70,75,* |
| Ratinhos wilde‐type versus ratinhos wilde‐type+ARA versus ratinhos transgénicos com sobre‐expressão de AT2R versus ratinhos transgénicos com sobre‐expressão de AT2R+ARA versus ratinhos transgénicos com sobre‐expressão de AT2R+knockouts para o AT1R41 |
| Administração de C21 em ratos (0,01, 0,03, 0,3mg/kg por dia)44 |
| Ratinhos com C21 (0,3mg/kg por dia) versus ratinhos com ARA versus ratinhos sem tratamento45 |
Na verdade, os estudos referenciados como70 e75 não estudam os efeitos do AT2R, mas sim os efeitos do bloqueio do AT1R com ARA. Porém, os autores concluem que parte dos efeitos dos ARA se deveram ao aumento da ativação do AT2R, porque este se encontrava sobre‐expressado. De toda a forma, os estudos ganhariam muito se incluíssem um grupo direcionado para o estudo do AT2R, como um duplo knockout, um transgénico que sobre‐expressasse AT2R ou um agonista direto do AT2R.
No primeiro estudo feito com o C21 nesta área, Kaschina44 concluiu que a estimulação do AT2R contribui para o melhoramento da função sistólica e diastólica no pós‐enfarte. Para além disso, também se verificou a ativação de mecanismos anti‐inflamatórios que contribuem para a proteção cardíaca. Porém, mesmo com a descoberta do C21, a controvérsia mantém‐se. Um estudo mais recente, de Jehle et al.45, conclui que, após EM, a estimulação direta do AT2R não melhora a remodelação cardíaca do ventrículo esquerdo, tendo até efeitos deletérios deletérios, nomeadamente o aumento do volume telessistólico e telediastólico no ventrículo esquerdo (comparativamente ao grupo tratado com um ARA).
Efeitos do recetor tipo 2 da angiotensina nas reações inflamatóriasA Ang II é considerada um agente pró‐inflamatório. É através da ligação ao AT1R que se dá o estímulo da resposta inflamatória. A disfunção endotelial é caracterizada pela diminuição da vasodilatação dependente do endotélio, que é precedida por uma série de alterações estruturais na parede vascular46. Verifica‐se aumento das moléculas adesivas, aumento do número de monócitos e da sua adesão ao endotélio, aumento da inflamação e aumento das fibras musculares lisas vasculares46,47. Isto pode levar à progressão de patologias como a aterosclerose, que provocam uma grande diminuição da capacidade vasodilatora dos vasos. A principal alteração que tem este efeito limitante é a diminuição da disponibilidade de NO46.
Contudo, o AT2R tem sido indicado como tendo efeitos anti‐inflamatórios. Sales et al.47 demonstraram um aumento da expressão de AT2R no local da lesão aterosclerótica. Ratinhos knockout para o AT2R apresentaram aumento do número de macrófagos, do número de células musculares lisas vasculares e da quantidade de colagénio nas placas. Verificaram também que a expressão AT2R aumenta num estado mais avançado da lesão, de forma a regular a composição da placa, limitando principalmente a acumulação de colagénio. Este AT2R exerce o seu efeito protetor através de: aumento do NO, diminuição do stress oxidativo, diminuição da proliferação celular e aumento da apoptose dos macrófagos.
Rompe et al.22 contribuíram para a determinação da via de sinalização da reação inflamatória ativada pelo AT2R. Também concluíram que o C21 reduz a adesão entre macrófados e inibe o NF‐kB. Outro estudo concluiu que a maioria dos ARAS disponíveis não são eficazes na inibição desta via inflamatória, porque esta também pode ser ativada pelo fator de necrose tumoral alfa (TNF‐α)48. O único ARA que se mostrou adequado foi o telmisartan porque pode ativar o peroxisome proliferator‐activated receptor (PPAR‐γ) que está associado à diminuição expressão de IL‐6 por estímulo da TNF‐α48.
Efeitos do recetor tipo 2 da angiotensina no rimO papel extenso que o AT2R tem a nível do rim ultrapassa os objetivos desta revisão. Assim, são apenas mencionadas os efeitos que estão diretamente envolvidos com a função cardiovascular. A nível do rim, o AT2R está expresso em altas quantidades no feto e em quantidades residuais no adulto. Mutações do gene do AT2R estão associadas ao desenvolvimento de anomalias congénitas do rim e trato urinário49. Por outro lado, a ausência de AT2R está associada a patologias renais, como o desenvolvimento de nefropatia diabética50.
A inibição do AT1R promove a natriurese, efeito este que se extingue quando se adiciona um inibidor do AT2R51. Assim, um dos efeitos do AT2R é a promoção da natriurese. Contudo, a ligação direta da Ang II ao AT2R não promove a natriurese51. A Ang II tem que ser convertida, primeiramente, em Ang III e esta sim, ao ligar‐se ao AT2R promove a natriurese52. A conversão da Ang II para Ang III é feita pela aminopeptidase A (APA)53. Esta Ang III pode ser convertida em Ang IV pela aminopeptidase N (APN)53. Assim, na presença de um inibidor do AT1R, quando há administração de Ang II concomitantemente com um inibidor da APN (PC‐18), há promoção da natriurese, pois mais Ang III se liga ao AT2R52. A situação contrária verifica‐se quando há administração de Ang II e de um inibidor da APA (EC‐33), pois haverá menos Ang III, logo menos natriurese52 (ver Figura 3). A natriurese promovida pelo AT2R está relacionada com a ativação do sistema quinina/NO/cGMP no túbulo renal proximal53. A nível do ramo ascendente espesso da ansa de Henle, também há promoção da natriurese, a partir da ativação do AT2R, apesar de não se saber se esta é por ligação da Ang III54. O mecanismo subjacente também envolve a produção de NO que inibe o cotransportador Na+/K+/2Cl[–], aumentando assim a excreção de sódio54.
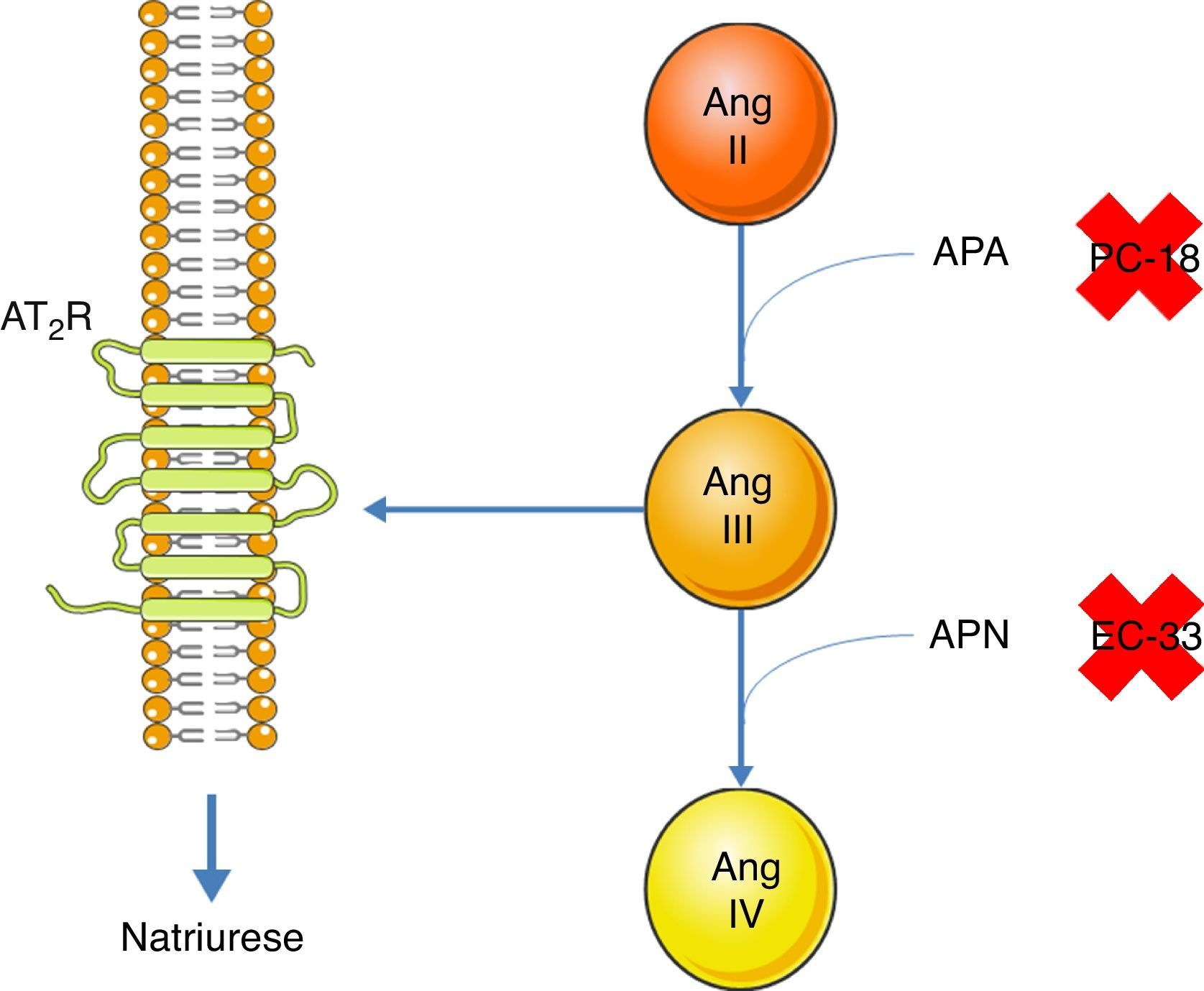
Promoção da natriurese via ativação do AT2R pela Ang III. A Ang II, para promover a natriurese, tem que ser convertida, primeiramente, em Ang III pela APA. Uma vez formada, esta Ang III pode ser convertida, pela APN, em Ang IV, que não tem a capacidade de promover a natriurese. A APA pode ser inibida pela PC‐18 e a APN pela EC‐33.
Legenda: Ang II: angeotensin II; Ang III: angeotensin III; Ang IV: angeotensin IV; APA: aminopeptidase A; APN: aminopeptidase N; AT2R: angiotensin receptor type 2. Os componentes das ilustrações são retirados do website: http://www.servier.co.uk/medical‐art‐gallery/.
Mais uma vez, os recetores da Ang II têm efeitos opostos num sistema. Neste caso, o seu principal papel passa pela regulação do sistema nervoso simpático, sendo que o AT1R o excita e o AT2R o inibe. Sendo assim, é percetível que ratinhos knockout para o AT2R tenham uma elevação na pressão sanguínea20. Os recetores que controlam este tónus simpático estão localizados no núcleo ventrolateral rostral do bolbo raquidiano. O AT2R regula a libertação de noradrenalina, reduzindo‐a20.
Alguns ligandos (GIP) do recetor tipo 2 da angiotensinaO AT2receptor interacting protein (ATIP)55 liga‐se seletivamente ao terminal carboxilo do AT2R e não se liga ao AT1R. A sua função é cooperar com o AT2R na inibição dos recetores cínases de tirosina independentes da proteína G. Por exemplo, inibe a ativação da ERK2, pelo epidermal growth factor receptor (EGFR), complementando assim a ação do AT2R na promoção da apoptose. É de referir que o ATIP não precisa que o AT2R seja ativado para se ligar a ele.
O AT2receptor binding protein of 50 kDa (ATBP50)56 é uma proteína de endereçamento do Golgi que regula do AT2R até à membrana celular, através da ligação ao seu terminal carboxilo. Assim, a ausência de ATBP50 promove a diminuição dos efeitos do AT2R.
A promyelotic zinc finger protein (PLZF), ao ligar‐se ao terminal carboxilo, estabelece uma nova via de sinalização para o AT2R, descrita por Senbonmatsu et al.57 (ver Figura 4). Após a ativação do AT2R pela Ang II, a PLZF liga‐se a este recetor e ambos são translocados até ao núcleo. O AT2R fica numa zona perinuclear e a PLZF entra para o núcleo, onde promove a transcrição do cínase PI3K‐p85α. Esta última está associada ao aumento da síntese proteica e à promoção do crescimento dos cardiomiócitos. Esta via pode ser uma possível explicação para os resultados contraditórios da função do AT2R a nível da hipertrofia cardíaca.
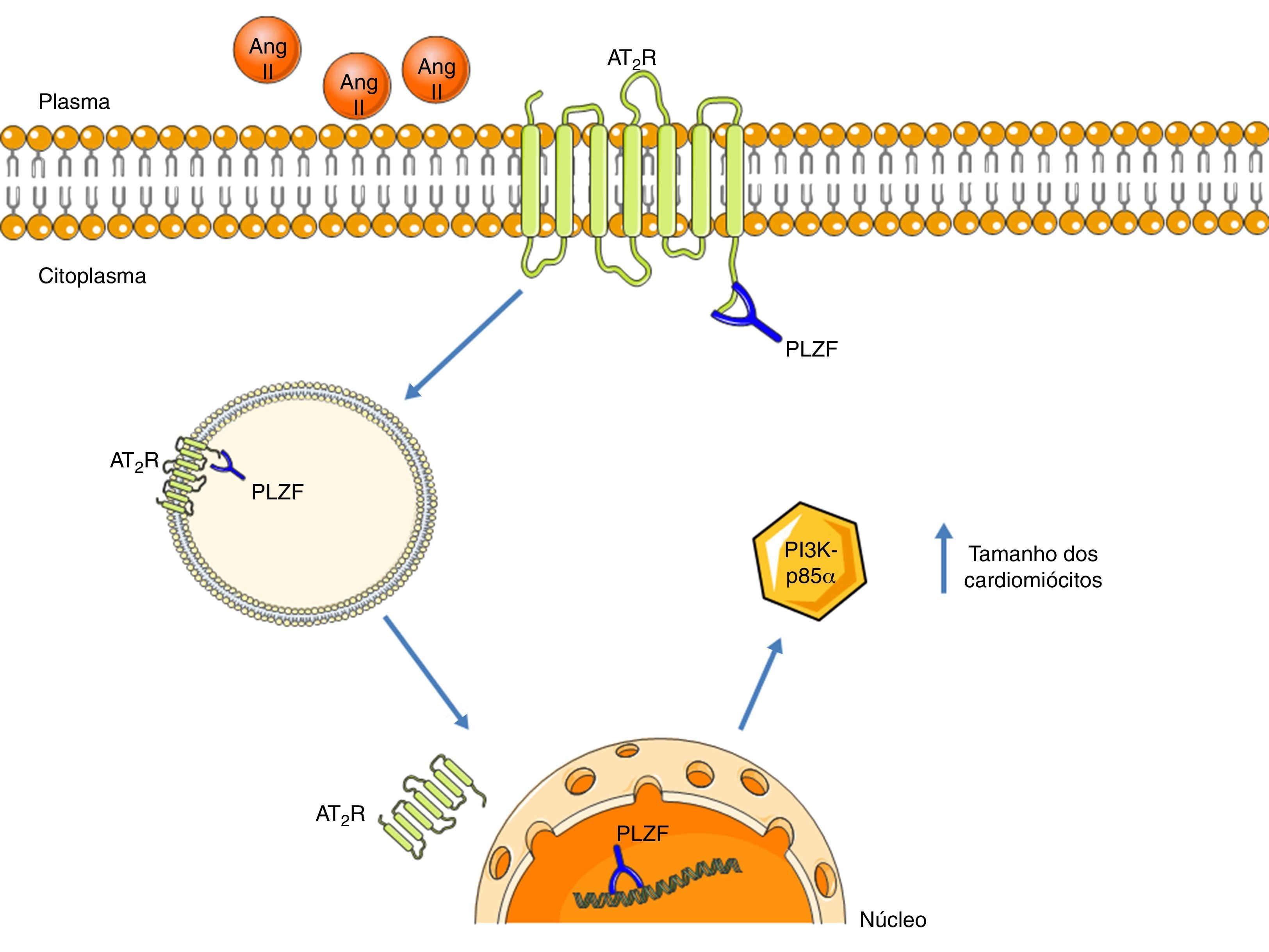
Via de sinalização do AT2R, através da ativação do PLZF. Aquando da ativação do AT2R, o PLZF liga‐se ao seu terminal carboxilo e promove a endocitose de ambos. A vesícula é translocada até ao núcleo, onde o PLZF entra, enquanto o AT2R permanece numa zona perinuclear. O PLZF promove a transcrição da PI3K‐p85α, que está associada à hipertrofia cardíaca.
Legenda: Ang II: angeotensin II; AT2R: angiotensin receptor type 2; PI3K‐p85α: phosphatidylinositol‐3 kinase‐p85α; PLZF: promyelotic zinc finger protein. Os componentes das ilustrações são retirados do website: http://www.servier.co.uk/medical‐art‐gallery/.
Na abordagem ao SRAA e às patologias associadas, temos duas grandes classes de fármacos: os ARA e os IECA. Os ARA são inibidores dos recetores da Ang II e os IECA são inibidores da enzima de conversão da angiotensina (ECA). Perante todos os efeitos benéficos do AT2R que têm vindo a ser estudados, foi sugerido que os ARA poderiam ter um melhor potencial terapêutico. Isto porque, se forem usados os IECA, a concentração da Ang II diminui, diminuindo a atividade do AT1R, mas também do AT2R. Já se forem usados os ARA, podemos inibir seletivamente o AT1R e manter, assim, a atividade benéfica do AT2R. Poder‐se‐á acrescentar ainda à terapêutica um agonista do AT2R.
É importante referir que existem dois tipos de ARA: os antagonistas competitivos e os agonistas inversos. Os agonistas inversos deverão ser preferidos, pois estabilizam o recetor na conformação inativa, desviando o equilíbrio da conformação ativa. Assim também impede a ativação do AT1R por stress mecânico (independente de ligando)23.
Apesar do tratamento com ARA parecer, teoricamente, mais eficaz, este ainda está longe de ser validado como primeira linha de escolha. Ma C et al.58 realizaram um estudo no qual compararam o tratamento de doentes hipertensos com IECA ou com ARA. Verificou‐se que os doentes tratados com os IECA têm uma melhor prevenção no surgimento de patologias como o enfarte agudo do miocárdio, fibrilação auricular e angina de peito instável. Assim, os autores concluíram que os IECA devem ser a primeira escolha para a prevenção destes riscos cardiovasculares. Por outro lado, a combinação de ambos os tratamentos não mostra vantagens na redução da mortalidade em doentes com insuficiência cardíaca, comparativamente ao tratamento só com IECA59. Esta combinação pode apresentar até alguns efeitos adversos como a hipercaliémia, hipotensão e insuficiência renal59,60.
A inibição da ECA1 está associada a uma maior atividade da ECA2 que forma Ang (1‐7), que ao ligar‐se ao recetor Mas produz efeitos protetores cardiovasculares61. Para além disto, como os IECA impedem a degradação da bradiquinina, esta poderá exercer os seus efeitos benéficos como por exemplo a vasodilatação (como já foi referido). Este é o principal aspeto que justifica a melhor eficácia dos IECA, no entanto, o acumulo de bradiquinina poderá estar relacionado com os seus efeitos adversos. De entre estes efeitos destaca‐se a angioedema (um edema limitado à derme e à submucosa)62,63. Em pacientes intolerantes a IECA há indicação para medicação com ARA64.
Existem, atualmente, oito ARA disponíveis no mercado: azilsartan, candesartan, eprosartan, irbesartan, losartan, olmesartan, telmisartan e valsartan65. Estes ARA apresentam diferenças farmacológicas que levam a diferentes escolhas para diferentes doentes. Por exemplo, o telmisartan é o que tem mais afinidade para o AT1R, o que tem maior semivida e o que é mais lipofílico, o que facilita a administração oral e a penetração nos tecidos66. É de referir que os ARA têm outros potenciais terapêuticos que transcendem o seu efeito no S‐RAA. Neste âmbito, o telmisartan mostra poder ativar o PPAR‐γ e assim ter também um efeito antidiabético67 (para além do anti‐inflamatório já referido em cima).
Csaba Farsang65 compilou uma lista interessante de recomendações para a administração de ARA em diferentes casos. Assim, para pacientes hipertensos ou normotensos, mas com elevado risco cardiovascular (p.ex. aterosclerose) o telmisartan está indicado por ter efeitos protetores que vão para além do controlo da pressão sanguínea. Por outro lado, em pacientes hipertensos com fatores de risco específicos são recomendados outros ARA, como o losartan (na prevenção de acidente vascular cerebral em indivíduos com hipertrofia ventricular esquerda e na prevenção da nefropatia diabética) ou o irbesartan (na prevenção da nefropatia diabética). Já em pacientes com um risco cardiovascular ainda mais elevado, nomeadamente numa insuficiência cardíaca ou numa disfunção ventricular esquerda, o candesartan, losartan ou valsartan são indicados.
Por vezes, associado aos IECA ou aos ARA são administrados outros fármacos para contrariar as consequências das patologias cardiovasculares. É importante o ter cuidado com estas combinações, pois delas podem surgir efeitos indesejáveis. Por exemplo, Lapi et al.68 descreveram que o triple whammy, a combinação de diuréticos com IECA ou ARA e com anti‐inflamatórios não‐esteroides, aumenta o risco de insuficiência renal aguda.
A descoberta do C21 trouxe uma nova via terapêutica. O uso clínico deste novo composto ainda não foi validado, mas os estudos que têm surgido fazem crer que tenha um forte potencial, principalmente na prevenção da lesão de órgãos decorrente da hipertensão. Na abordagem à inflamação, o C21 parece ser uma boa alternativa aos fármacos atuais que inibem a IL‐6 e o TNF‐α, pois estes têm grandes efeitos adversos22. Considerando que o AT2R se expressa em baixa quantidade nos tecidos saudáveis e em alta quantidade nos tecidos lesados, a terapia com C21seria direcionada para o sítio da lesão, limitando possíveis efeitos adversos só a esse local e não a nível sistémico69. Esta será, talvez, a maior vantagem do tratamento com C21.
ConclusãoO AT2R tem sido alvo de grande controvérsia desde a sua descoberta. Resultados opostos tornaram as suas funções confusas e de difícil compreensão. Com a descoberta progressiva de novos intervenientes nas vias de sinalização e novas proteínas que se podem associar ao recetor, as funções do AT2R têm vindo a ser clarificadas. O que parece ser consensual é que tem um efeito oposto ao do AT1R na maioria dos tecidos. Se isso é suficiente para ter funções protetoras na patologia cardiovascular ainda é discutível. Novos estudos, principalmente com o agonista C21, são promissores para a dissipação das dúvidas sobre o AT2R. Num futuro próximo, poderão surgir novas terapêuticas que incluam a estimulação dos efeitos protetores do AT2R, de forma a que sejam minimizadas as consequências de patologias cardiovasculares como a aterosclerose, o EM ou a insuficiência cardíaca. Assim, o «mundo do AT2R» permanece como uma janela aberta para uma nova e melhorada ação médica.
Conflito de interesesOs autores declaram não haver conflito de interesses.
