



The angiotensin type 2 receptor, AT2R, has been described as having opposite effects to the angiotensin type 1 receptor, AT1R. Although the quantities of the AT2R found in the adult are low, its expression rises in pathological situations. The AT2R has three major signaling pathways: activation of serine/threonine phosphatases (promoting apoptosis and antioxidant effects), activation of the bradykinin/NO/cGMP pathway (promoting vasodilation), and activation of phospholipase A2 (associated with regulation of potassium currents). The AT2R appears to have effects in vascular remodeling, atherosclerosis prevention and blood pressure lowering (when associated with an AT1R inhibitor). After myocardial infarction, the AT2R appears to decrease infarct size, cardiac hypertrophy and fibrosis, and to improve cardiac function. However, its role in the heart is controversial. In the kidney, the AT2R promotes natriuresis. Until now, treatment directed at the renin–angiotensin–aldosterone system has been based on angiotensin-converting enzyme inhibitors or angiotensin type 1 receptor blockers. The study of the AT2R has been revolutionized by the discovery of a direct agonist, C21, which promises to become part of the treatment of cardiovascular disease.
O recetor da angiotensina do tipo 2, AT2R, tem vindo a ser descrito como tendo ações opostas ao recetor da angiotensina do tipo 1, AT1R. Apesar do AT2R existir em baixas quantidades no adulto, a sua expressão sobe bastante em situações patológicas. O AT2R tem três grandes vias de sinalização: a ativação fosfátases de serina/treonina (promoção da apoptose celular e efeitos antioxidantes); ativação da via bradiquinina/NO/cGMP (promoção de vasodilatação) e ativação da fosfolípase A2 (associada ao controlo das correntes de potássio). O AT2R parece ter um efeito na remodelação vascular, na prevenção da aterosclerose e na descida da pressão sanguínea (quando associada a um inibidor do AT1R). Após enfarte do miocárdio, o AT2R parece diminuir o tamanho do enfarte, a hipertrofia cardíaca, a fibrose e aumentar a função cardíaca. Contudo, o seu papel a nível cardíaco é o mais controverso. A nível renal o AT2R promove a natriurese. Até agora, a terapêutica direcionada para o sistema renina-angiotensina-aldosterona é à base de inibidores da enzima de conversão da angiotensina (IECA) ou de inibidores do recetor da angiotensina tipo 1 (ARA). O estudo do AT2R foi revolucionado pela descoberta de um agonista direto, o C21, que promete integrar parte da terapêutica das doenças cardiovasculares.
In the renin–angiotensin–aldosterone system (RAAS), the most widely studied angiotensin (Ang) II receptor is the type 1 receptor, AT1R. This receptor is responsible for most of the effects of RAAS activation, including vasoconstriction, sodium retention, aldosterone release, cell proliferation, cardiac and vascular hypertrophy, and modulation of oxidative stress and inflammation. Ang II also binds to another receptor, AT2R, which was discovered over 20 years ago.1 However, its functions are still not fully understood. Activation of the AT2R has been reported as having opposite effects to that of the AT1R,2–5 and thus appears to have a protective effect in conditions such as hypertension, atherosclerosis and myocardial infarction (MI). Both the AT1R and the AT2R are G protein-coupled receptors (GPCRs)6 with 34% sequence homology.7 Various GPCR-interacting proteins (GIPs) also interact with these receptors, binding to the C-terminus.8,9 In adults, the AT1R is expressed ubiquitously, while the AT2R is found in low quantities, mainly in the blood vessels, kidneys, adrenal medulla, uterus and ovaries, heart, and certain brain nuclei.2,10 However, its expression rises in the above pathological situations. The physiological role of the AT2R in adults is thus insignificant, but in the fetus, the opposite is seen: the AT2R is more abundant, which may be related to its function in general physiological development.
The aim of this review is to describe the functions of the AT2R in the cardiovascular system and to investigate their role in possible treatments.
Signal transduction pathways of the AT2RThe Ang II receptors form homodimers and heterodimers between each other. The homodimers AT1R/AT1R and AT2R/AT2R strengthen the effects of their respective receptors.9 Formation of the heterodimer AT1R/AT2R reduces the signaling of the AT1R, one way in which the AT2R directly inhibits the effects of the AT1R.11 The AT2R also forms heterodimers with the bradykinin B2 receptor (B2R), increasing production of nitric oxide (NO).12 This interaction is important in the kinin/NO/cGMP system.
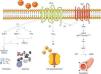
Once activated, the AT2R has three main signaling pathways: activation of serine/threonine phosphatases, activation of the bradykinin/NO/cGMP pathway, and activation of phospholipase A213 (Figure 1).
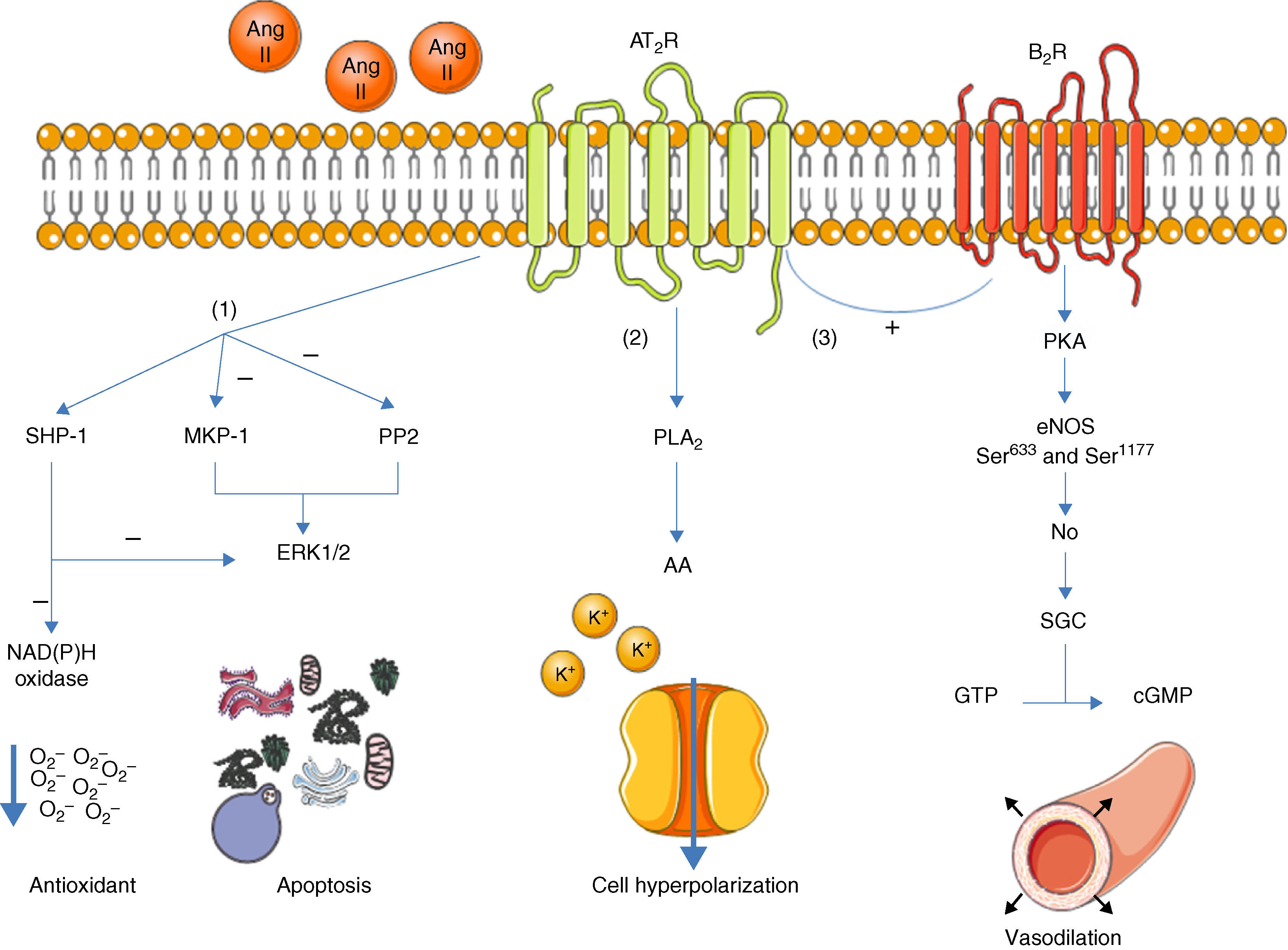
Main signaling pathways of the AT2R. Activation of the receptor leads to three pathways: activation of serine/threonine phosphatases (1); activation of phospholipase A2 (2); and activation of the bradykinin/NO/cGMP pathway (3). AA: arachidonic acid; Ang II: angiotensin II; AT2R: angiotensin receptor type 2; B2R: bradykinin receptor B2; cGMP: cyclic guanosine monophosphate; eNOS: endothelial nitric oxide synthase; ERK1/2: extracellular-regulated kinase 1 and 2; GTP: guanosine triphosphate; MKP-1: MAP kinase phosphatase; NO: nitric oxide; PKA: protein kinase A; PLA2: phospholipase A2; PP2: protein phosphatase 2; sGC: soluble guanylate cyclase; SHP-1: SH2 domain-containing tyrosine phosphatase.
With regard to phosphatases, the role of the AT2R has been studied in the activation of MAP kinase phosphatase (MKP-1), protein phosphatase 2 (PP2A) and SH2 domain-containing tyrosine phosphatase (SHP-1).3,13
Activation of MKP-1 and PP2A by the AT2R results in inhibition of ERK1/2 (ERK1/2), inducing apoptosis. When activated, SHP-1 inhibits ERK1/2 and NAD(P)H oxidase (stimulated by the AT1R14), and is thus part of the endothelium's oxidative stress defense.15
Activation of the bradykinin/NO/cGMP pathway is associated with vasodilation.4,16 When activated, the AT2R stimulates the B2R receptor, which in turn induces phosphorylation of endothelial nitric oxide synthase (eNOS) at Ser633 and Ser1177 via a PKA-mediated signaling pathway.17 NO production is thus increased, activating guanylate cyclase (sGC), which synthesizes cGMP from GTP, in turn promoting vasodilation. Although these mechanisms have been described before,16,18 the role of the AT2R in regulation of blood pressure (BP) is still the subject of debate.1
The AT2R also stimulates phospholipase A2 (PLA2) activity and arachidonic acid (AA) formation; the latter regulates potassium currents and can lead to cell hyperpolarization and reduced excitability.3,19 This effect appears to be particularly important in reducing sympathetic activity.20
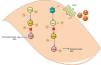
Guilluy et al.21 described an alternative signaling pathway to the bradykinin/NO/cGMP system in vascular smooth muscle cells that culminates in phosphorylation and inactivation of RhoA on Ser188, leading to vasodilation (Figure 2). Ste20-related kinase (SLK) is responsible for this phosphorylation, which is independent of eNOS, PKA and PKG. The effect is weakened when SLK is phosphorylated, hence SLK phosphorylates RhoA when in the dephosphorylated state. Meanwhile, casein kinase 2 (CK2) is responsible for the basal phosphorylation of SLK. The role of the AT2R in this pathway is thus to reduce the activity of CK2 in order to increase the quantity of dephosphorylated SLK. Since CK2 is active when phosphorylated, the AT2R achieves its inhibitory effect through SHP-1, which dephosphorylates CK2.
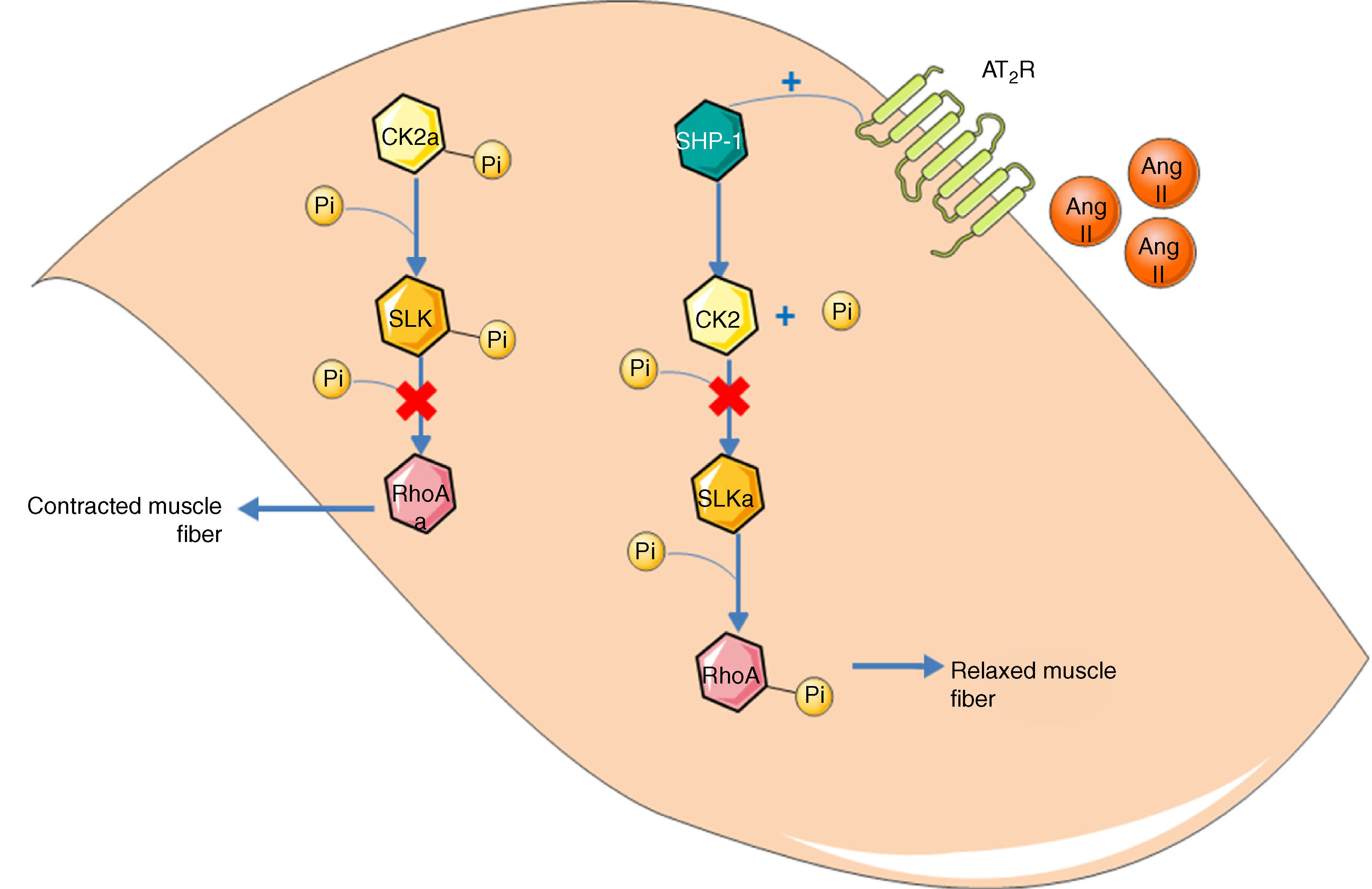
Alternative vasodilation pathway mediated by the AT2R. Activation of the AT2R activates SHP-1, which dephosphorylates and inactivates CK2; the latter no longer phosphorylates (and inactivates) SLK. Activated SLK phosphorylates and inactivates RhoA, leading to relaxation of vascular smooth muscle fibers. Ang II: angiotensin II; AT2R: angiotensin receptor type 2; CK2: casein kinase 2; Pi: inorganic phosphate; RhoA: Ras homolog family member A; SHP-1: SH2 domain-containing tyrosine phosphatase; SLK: Ste20-related kinase; SLKa: activated SLK.
The AT2R is also involved in anti-inflammatory pathways that reduce the expression of interleukin-6 (IL-6),22 mainly in stimulation of serine/threonine phosphatases and in the synthesis of AA, which then produces the metabolite 11,12-EET, part of the inflammatory pathway. These two processes converge to reduce the activity of nuclear factor κB (NF-κB), which in turn reduces IL-6.22
It should be noted that these receptors can be activated independently of binding to Ang II. The AT1R may be activated solely by mechanical stress, leading to increased expression of calcineurin which, in turn, causes cardiac hypertrophy,23 while in a study of mouse ovary cells, Miura and Karnik24 showed that the AT2R does not need angiotensin to induce apoptosis. It is now accepted that some AT2Rs are constitutively active in the form of homo-oligomers.25 These characteristics of the Ang II receptors are particularly relevant in drug treatments, as discussed below.
The AT2R and its agonist C21The first studies on the AT2R were all indirect, some involving selective activation of the AT2R by supplying Ang II in the presence of an AT1R inhibitor II, and some using an AT2R antagonist such as PD123319 to study the effects of disabling this receptor. However, the latter approach has the disadvantage that PD123319 lacks specificity when applied at high doses.1 Other studies have used mice genetically engineered to under- or over-express the AT2R gene.
The most direct way to study the AT2R is to use a specific agonist. Until 2004 the best available agonist was CGP42112A, but this ligand is rapidly degraded in vivo and has antagonistic as well as agonistic properties.1 However, in that year, Wan et al.26 reported the design and synthesis of a new agonist, compound 21 (C21), which has revolutionized the study of the AT2R. This non-peptide agonist, which can be administered orally, allows direct in vitro and in vivo stimulation of the AT2R without affecting the AT1R.
Vascular effects of the AT2RAng II regulates various processes involved in vascular pathophysiology, including growth and apoptosis of vascular cells, migration of vascular smooth muscle cells, inflammatory responses and extracellular matrix remodeling.13 Ang II can bind to the AT1R or to the AT2R, which have opposite effects. For example, it has been shown that a deletion in the AT2R gene is associated with higher BP, reflecting the absence of AT2R-related vasodilation.4,27 A study by Savoia et al.28 showed, for the first time, upregulation of the AT2R in peripheral arteries of hypertensive diabetic patients treated with the AT2R blocker valsartan for one year. This increase in the AT2R was associated with lower blood pressure in these patients in a dose-dependent fashion. The authors confirmed this effect by exposing samples of precontracted peripheral arteries ex vivo to valsartan; the result was again increased AT2R expression and vasodilation. This increase was seen mainly in the media of the vessel wall, and so the authors hypothesize that the AT2R stimulates vasodilation directly in vascular smooth muscle cells via RhoA, as described above.
Zhang et al.29 studied the functions of the AT1R and the AT2R in the coronary circulation and concluded that stimulation of the AT1R evoked vasoconstriction and stimulation of the AT2R induced vasodilation. Removal of endothelium or administration of an NO synthase inhibitor abolished the vasodilatory response of the AT2R. The authors also found that superoxide production via AT1R activation of NAD(P)H oxidase, as described above, inhibited vasodilation, the superoxide reacting with NO and thus acting as an NO scavenger.
The AT2R is abundant in the fetus, and it has therefore been argued that it plays a role in normal vascular development and growth.3 AT2R expression is linked to fetal angiogenesis and influences the phenotype of vascular smooth muscle cells by altering ERK activity.30 AT2R expression peaks towards the end of gestation; this is linked to reduced DNA synthesis in the aorta.31 Normal vascular differentiation is thus achieved by the opposing actions of the AT1R and the AT2R.32 In adults, the AT2R continues to play a part in vascular remodeling, inducing apoptosis and inhibiting cell proliferation (which is promoted by the AT1R). This is particularly evident following carotid artery balloon injury, following which in-vivo transfer of the AT2R gene significantly attenuates neointimal formation.31,33 Dysregulation of this remodeling process is associated with pathological states. For example, loss of AT2R expression accelerates the development and rupture of aortic aneurysms in a mouse model of Marfan syndrome.34 In these mice, an AT1R inhibitor (the angiotensin receptor blocker [ARB] losartan) slowed aneurysm progression but full protection required intact AT2R signaling.34 There are also indications that the AT2R plays a part in protecting against vascular hypertrophy.35
Following the discovery of C21, new light has been shed on the vascular effects of the AT2R. One idea – that AT2R-related vasodilation reduces BP – has been disproved by new research: administration of C21 does not by itself reduce BP in either normotensive36,37 or hypertensive animals.36,38,39 The only study to show a fall in BP was the one that first described C21, in hypertensive rats,26 and furthermore the animals were anesthetized, which would make them much more sensitive to pharmacological modulation of the RAAS.1 Since the predominance of the AT1R hinders investigation of the effects of direct AT2R stimulation in vivo, Bosnyak et al. administered C21 together with a low dose of the ARB candesartan, which does not lower BP by itself, and saw a significant BP fall in hypertensive rats. This response was abolished by administration of an AT1R antagonist.36 When a high-dose ARB is added to C21 there is no additional effect on BP beyond that of the ARB alone.38,39
These studies with C21 confirm that the AT2R mainly confers protection against vascular injury. Rehman et al.38 showed in hypertensive rats that C21 alone reduces vascular stiffness, aortic medial collagen content and the coronary perivascular collagen:media cross-sectional area ratio. Paulis et al.39 studied the effects of C21 independently of NO, using an NO synthase inhibitor (l-NAME) in all experimental groups except controls; they concluded that chronic AT2R stimulation prevented aortic stiffening and collagen accumulation without preventing hypertension.
Effects of the AT2R on cardiac structureThe two Ang II receptors are also involved in remodeling in the heart, but there is disagreement as to the underlying processes. Under normal conditions the AT2R is expressed in the heart at very low levels, but in pathological conditions such as hypertension or MI its expression increases greatly, to the extent that its action predominates over that of the AT1R. Most of the controversy about the actions of the AT2R has arisen from studies of cardiac hypertrophy, fibrosis and post-MI cardiac function.1 Post-MI remodeling has an acute phase of infarct expansion, followed by hypertrophy and interstitial fibrosis of cardiomyocytes that did not undergo ischemia.40 Most studies indicate that the AT2R has a beneficial effect in the heart. The main findings of a review by Johren et al.,3 dating from 2004 (before the discovery of C21), of studies in AT2R knockout mice, were that in the myocardium, deletion of the AT2R gene leads to increased AT1R expression and thus AT1R-mediated cell proliferation, and increased cardiac hypertrophy, reduced survival and increased left ventricular (LV) dilatation after MI. Among the disagreements in this field, some studies indicate that knocking out the AT2R promotes perivascular fibrosis, while others come to exactly the opposite conclusion.
Other studies followed the review by Johren et al., but the controversy continued. Table 1 presents a summary of more recent research. Of particular note is Voros et al.,41 who compared five groups of mice and found that both blockade of the AT1R and overexpression of the AT2R had beneficial effects on cardiac remodeling, and the AT1R knockout transgenic mice with AT2R overexpression consistently had higher ejection fraction, smaller LV end-diastolic volume and less fibrosis in regions adjacent to the infarct. However, other studies contradict the idea that the AT2R has protective effects in the heart. In a study by Tschope et al.,42 AT2R knockout mice showed no differences from controls after MI in LV systolic and diastolic function or in fibrosis or remodeling of the extracellular matrix. The authors concluded that ischemia-induced hypertrophy and fibrosis are independent of the AT2R. Van Esch et al. showed that the absence of the AT2R did not enhance the effects of the AT1R, while in the absence of the AT1R, the cardiac effects mediated by the AT2R were not observed.
Effects of the AT2R after myocardial infarction and in heart failure.
| Beneficial effects after MI (compared to controls) |
| Reduced infarct size44,70 |
| Increased maximum and minimum dP/dt44,70,71 |
| Increased ejection fraction41,70–73 |
| Increased contractility44 |
| Reduced LV end-systolic volume41,72,73 |
| Reduced LV end-diastolic volume41,72,73 |
| Reduced LV end-diastolic pressure44,71 |
| Reduced left atrial pressure70a |
| Reduced LV hypertrophy74 |
| Reduced LV/body weight ratio74 |
| Reduced lung/body weight ratio74 |
| Reduced interstitial fibrosis in regions adjacent to the infarct41 |
| Reduced levels of monocyte chemoattractant protein-1, myeloperoxidase and interleukins (anti-inflammatory effects)44 |
| Absence of effects or deleterious effects after MI (compared to controls) |
| No changes in systolic or diastolic function42 |
| No changes in fibrosis and extracellular matrix42 |
| Increased LV end-systolic and end-diastolic volume45 |
| No inhibition of the effects of the AT1R43 |
| Beneficial effects in HF (compared to controls)a |
| Increased maximum and minimum dP/dt75 |
| Increased ejection fraction75 |
| Reduced mean pulmonary artery pressure75 |
| Reduced LV end-systolic volume75 |
| Reduced LV end-diastolic volume75 |
| Reduced LV end-diastolic pressure75 |
| Reduced aldehydes (a marker of oxidative stress)75 |
| Study approach |
| AT2R knockout mice42 |
| Transgenic mice overexpressing AT2R vs. controls72,73 |
| Wild-type mice+ARB vs. AT2R knockout mice+ARB74 |
| AT1R knockout mice+AT2R wild-type mice vs. AT2R knockout mice+AT1R wild-type43 |
| AT2R promoter injected via an adenoviral vector into left ventricle of rats71 |
| AT1R blockade with ARBs in dogs70,75a |
| Wild-type mice vs. wild-type mice+ARB vs. transgenic mice overexpressing AT2R vs. transgenic mice overexpressing AT2R+ARB vs. transgenic mice overexpressing AT2R and AT1R knockout41 |
| Treatment with C21 in rats (0.01, 0.03, 0.3 mg/kg/day)44 |
| Mice treated with C21 (0.3 mg/kg/day) vs. treated with ARB vs. untreated controls45 |
ARB: angiotensin receptor blocker; HF: heart failure; MI: myocardial infarction.
Refs. 70 and 75 did not study the effects of the AT2R, but of AT1R antagonism with ARBs. However, the authors concluded that part of the effects of the ARBs were due to increased AT2R activation since this was overexpressed. Studies would benefit from the inclusion of a group specifically designed to study the AT2R, such as double knockout mice, transgenic mice overexpressing the AT2R, or a direct AT2R agonist.
The first study with C21 in this area, by Kaschina et al.,44 concluded that AT2R stimulation may improve post-MI systolic and diastolic function, and also observed activation of anti-inflammatory mechanisms that afford some degree of cardiac protection. However, even after the discovery of C21, the controversy continues; a more recent study by Jehle et al.45 concluded that direct stimulation of the AT2R does not attenuate post-MI LV remodeling and in fact has adverse effects, particularly increased LV end-systolic and end-diastolic volume, compared to the group treated with an ARB.
Effects of the AT2R on the inflammatory responseAng II is considered a proinflammatory agent; binding to the AT1R triggers an inflammatory response. Endothelial dysfunction is characterized by impaired endothelium-dependent vasodilation, which is preceded by a series of structural changes in the vessel wall,46 including increases in adhesion molecules, number of monocytes and their adhesion to the endothelium, inflammation, and vascular smooth muscle fibers.46,47 These changes are linked to the progression of pathological conditions such as atherosclerosis, which are associated with a sharp decline in vasodilatory capacity, the main factor in which is reduced NO availability.46
However, it has been suggested that the AT2R has anti-inflammatory effects. Sales et al.47 demonstrated increased AT2R expression at the site of atherosclerotic lesions. AT2R knockout mice had increased macrophages, vascular smooth muscle cells and collagen in plaques, and increased AT2R expression was observed in more advanced stages of lesion evolution, in which it appears to play a part in regulation of plaque composition, mainly by inhibiting collagen accumulation. Thus the AT2R exerts its protective effect by increasing NO, reducing oxidative stress and cell proliferation, and promoting macrophage apoptosis.
Further contributing to the study of inflammatory signaling pathways activated by the AT2R, Rompe et al.22 concluded that C21 reduces adhesion between macrophages and inhibits NF-κB. According to another study, most currently available ARBs are ineffective in inhibiting this inflammatory pathway because it is also activated by tumor necrosis factor α (TNF-α).48 The only effective ARB is telmisartan, which activates peroxisome proliferator-activated receptor γ (PPAR-γ), which reduces TNF-α-induced IL-6.48
Effects of the AT2R on the kidneyThe extensive role of the AT2R in the kidney is beyond the scope of this review, and so only those effects that are directly related to cardiovascular function will be described.
The AT2R is expressed in the kidney at high levels in the fetus but at very low levels in the adult. Mutations in the AT2R gene are associated with congenital abnormalities of the kidney and urinary tract,49 while absence of the receptor is linked to renal diseases such as diabetic nephropathy.50
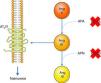
Inhibition of the AT1R promotes natriuresis, an effect that is abolished when an AT2R blocker is administered,51 demonstrating that natriuresis is one of the effects of the AT2R. However, direct binding of Ang II to the AT2R does not have this effect51; Ang II must first be converted to Ang III, which then binds to the AT2R, resulting in natriuresis.52 Ang II is converted to Ang III by aminopeptidase A (APA), while Ang III is converted to Ang IV by aminopeptidase N (APN).53 Thus, in the presence of an AT1R blocker, administration of Ang II together with PC-18, a specific inhibitor of APN, results in natriuresis, since more Ang III binds to the AT2R.52 The opposite occurs when Ang II is administered with EC-33, a selective APA inhibitor, which reduces Ang III and hence natriuresis52 (Figure 3). AT2R-mediated natriuresis occurs via activation of the kinin/NO/cGMP system in the renal proximal tubule.53 Natriuresis is also triggered by AT2R activation in the thick ascending limb of Henle, although it is not known whether this is due to binding to Ang III.54 The underlying mechanism also involves the production of NO, which inhibits the Na+/K+/2Cl− cotransporter, thus increasing sodium excretion.54
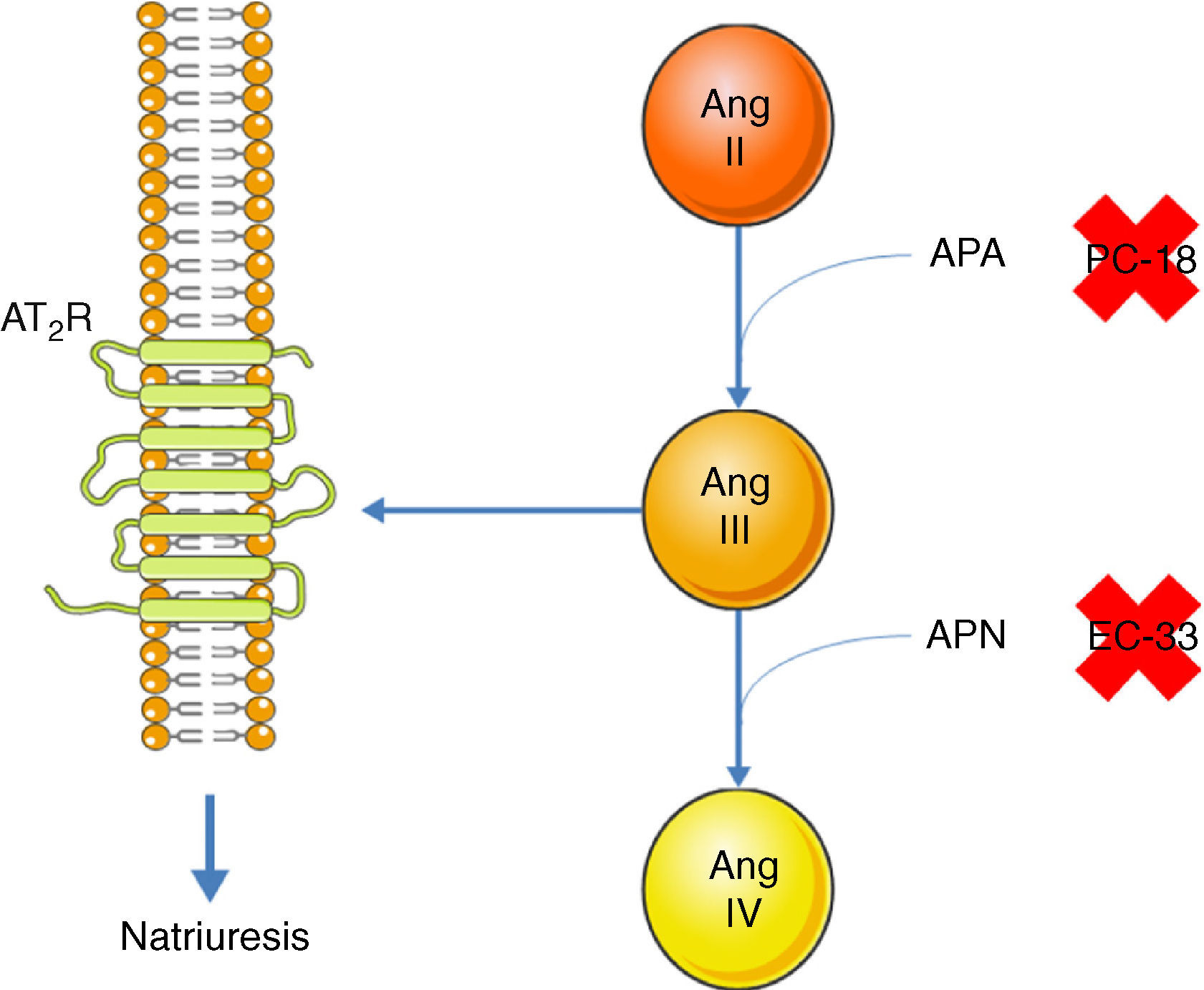
Promotion of natriuresis via activation of the AT2R by Ang III. Ang II does not promote natriuresis until it has been converted to Ang III by APA. Ang III is then converted by APN to Ang IV, which does not trigger natriuresis. APA can be inhibited by PC-18 and APN by EC-33. AT2R: angiotensin receptor type 2; Ang II: angiotensin II; Ang III: angiotensin III; Ang IV: angiotensin IV; APA: aminopeptidase A; APN: aminopeptidase N.
The Ang II receptors also have opposing effects in the nervous system. Their main role is in regulating the sympathetic nervous system, which the AT1R stimulates and the AT2R inhibits. It is thus logical that AT2R knockout mice should have higher BP.20 The receptors that control sympathetic tone are located in the rostral ventrolateral medulla, where AT2R overexpression reduces noradrenaline excretion.20
AT2R ligands (GPCR-interacting proteins)AT2 receptor-interacting protein (ATIP)55 binds selectively to the C-terminus and does not bind to the AT1R. It cooperates with the AT2R to transinactivate receptor tyrosine kinases that are independent of G proteins. For example, it inhibits activation of epidermal growth factor receptor (EGFR)-induced ERK2, thus complementing the proapoptotic effects of the AT2R. The AT2R does not need to be activated for ATIP to bind to it.
AT2R binding protein of 50 kDa (ATBP50)56 is a Golgi membrane-associated protein that regulates the transport of the AT2R to the cell membrane by binding to its C-terminus. Downregulation of ATBP50 decreases the antiproliferative effects of the receptor.
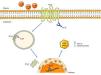
Promyelocytic zinc finger protein (PLZF) provides another signaling mechanism for the AT2R, binding to its C-terminus, as described by Senbonmatsu et al.57 (Figure 4). After activation by Ang II, the AT2R binds to PLZF and both translocate to the nucleus. The AT2R remains in the perinuclear region while PLZF enters the nucleus, where it promotes transcription of the kinase PI3K p85α, which is associated with increased protein synthesis and cardiomyocyte growth. This pathway may explain the contradictory findings concerning the role of the AT2R in cardiac hypertrophy.
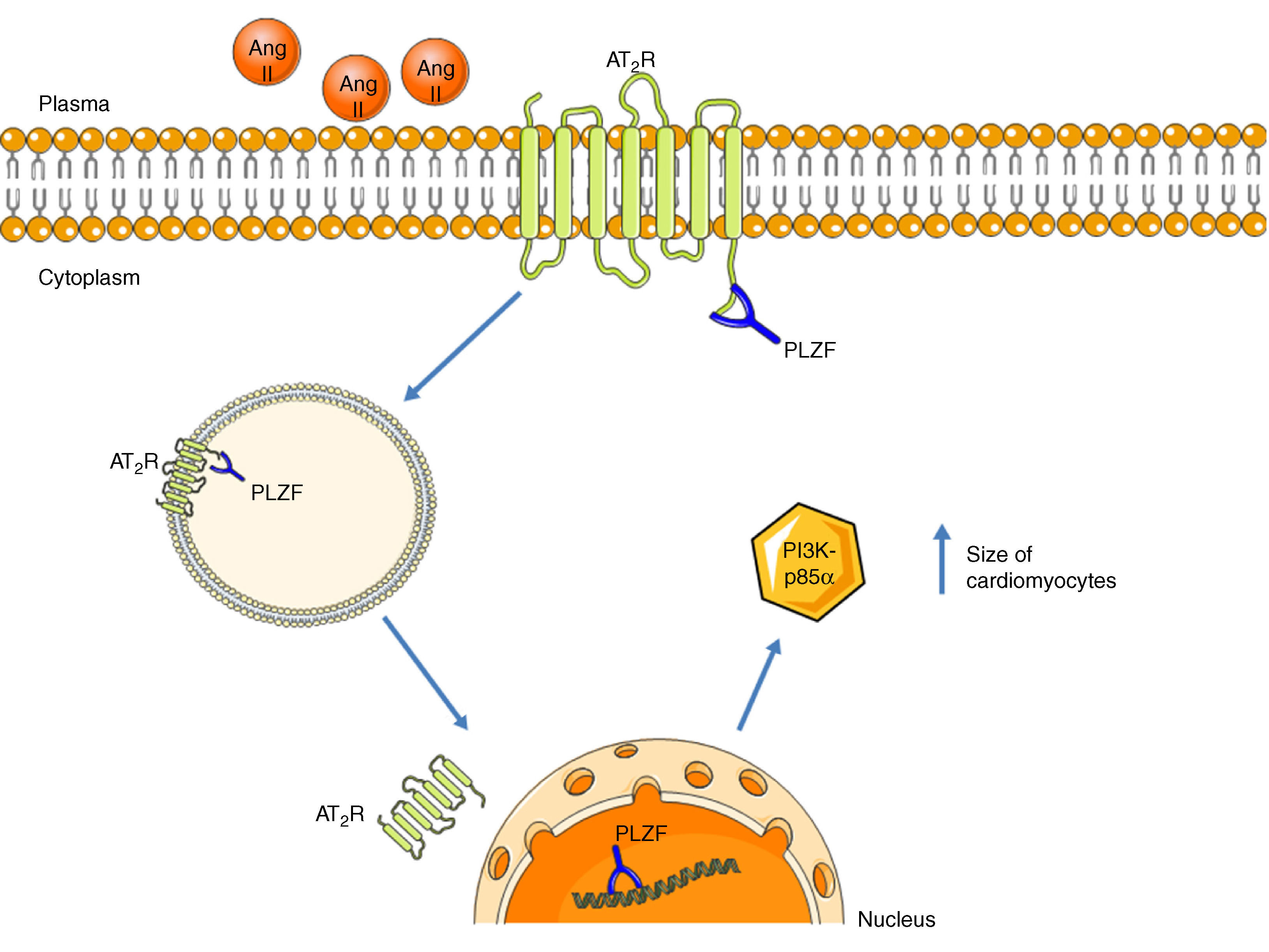
AT2R signaling pathway via activation of PLZF. Following activation of the AT2R, PLZF binds to its C-terminus and both undergo endocytosis. The vesicle translocates to the nucleus, which PLZF enters, while the AT2R remains in the perinuclear region. PLZF promotes the transcription of PI3K-p85α, which is associated with cardiac hypertrophy. AT2R: angiotensin receptor type 2; Ang II: angiotensin II; PLZF: promyelocytic zinc finger protein; PI3K p85α: phosphatidylinositol-3 kinase-p85α.
The two main drug classes used for modulating the RAAS and treating related pathological conditions are ARBs and angiotensin-converting enzyme (ACE) inhibitors. It has been suggested, in view of the beneficial effects of the AT2R revealed by recent research, that ARBs have greater therapeutic potential than ACE inhibitors, since the latter reduce Ang II levels and thus the activity of the AT2R as well as the AT1R, whereas ARBs allows selective inhibition of the AT1R while maintaining the beneficial action of the AT2R. An AT2R agonist can also be added.
There are two types of ARB: competitive antagonists and inverse agonists. The latter are preferable since they stabilize the receptor in its inactive conformation, as well as preventing activation of the AT1R by ligand-independent mechanical stress.23
Although ARBs should theoretically be more effective, they are still far from being validated as the first-line choice. Ma et al.,58 comparing ACE inhibitors and ARBs in the treatment of elderly hypertensive patients, found that ACE inhibitors were more effective in preventing cardiovascular events including MI, atrial fibrillation and unstable angina, and concluded that ACE inhibitors should be the first choice for a spectrum of cardiovascular risks. Combining the two drugs does not reduce mortality in heart failure patients any more than ACE inhibitors alone,59 and is moreover associated with complications such as hyperkalemia, hypotension and renal failure.59,60
Inhibition of ACE1 is associated with greater activity of ACE2; the latter produces Ang 1–7, which binds to the Mas receptor and has cardioprotective effects.61 Furthermore, ACE inhibitors block the degradation of bradykinin, which has beneficial effects including vasodilation, as described above; this is the main reason for the greater efficacy of ACE inhibitors. However, the accumulation of bradykinin can have adverse effects, especially angioedema (edema of the dermis and submucosa).62,63 ARBs are an alternative for patients who do not tolerate ACE inhibitors.64
Eight ARBs are currently available: azilsartan, candesartan, eprosartan, irbesartan, losartan, olmesartan, telmisartan and valsartan,65 each with specific pharmacological characteristics and indications for different patients. For example, telmisartan has the greatest affinity for the AT1R and the longest half-life and is the most lipophilic, resulting in easier oral administration and penetration into tissue.66 It should be borne in mind that ARBs have effects other than RAAS modulation; telmisartan activates PPAR-γ and thus has an antidiabetic effect,67 as well as the anti-inflammatory properties described above.
Farsang65 compiled an interesting list of recommendations for the administration of ARBs in different cases. Thus, telmisartan is indicated for hypertensive patients and normotensive individuals with high cardiovascular risk (for example due to atherosclerosis) because of its cardioprotective effects, which go beyond BP control, while other ARBs are recommended for hypertensive patients with specific risk factors – losartan for prevention of stroke in patients with LV hypertrophy and for prevention of diabetic nephropathy, and irbesartan, also for prevention of diabetic nephropathy. For patients with even higher cardiovascular risk, such as those with heart failure or LV dysfunction, candesartan, losartan or valsartan are indicated.
Other drugs are sometimes added to ACE inhibitors or ARBs to treat cardiovascular disease. Caution must be exercised with such combinations, which can have deleterious effects. For instance, Lapi et al.68 describe the ‘triple whammy’ of the combination of diuretics, ACE inhibitors or ARBs, and non-steroidal anti-inflammatory drugs, which increases the risk of acute renal failure.
The discovery of C21 has opened up new therapeutic possibilities. This new compound has not yet been validated for clinical use, but studies suggest that it has considerable potential, particularly for the prevention of hypertension-related target organ damage and in the treatment of inflammation as an alternative to currently used drugs that inhibit IL-6 and TNF-α, which have significant adverse effects.22 Since the AT2R is expressed in low quantities in healthy tissue but in high quantities in damaged tissue, therapy with C21, and possible adverse effects, would be limited to the site of the injury, rather than being systemic.69 This could in fact be the main advantage of the new compound.
ConclusionThe AT2R has been the subject of considerable controversy since its discovery, with contradictory findings that make its actions difficult to fully understand. However, as more factors influencing its signaling pathways and more proteins that can bind to the receptor have been discovered, its functions are becoming clearer. There is now general agreement that it has opposite effects to the AT1R in most tissues, but whether this is sufficient to conclude that it has protective effects in cardiovascular disease is still debatable. New studies, particularly on the agonist C21, promise to help dispel the uncertainties concerning the AT2R. Novel therapies may soon be available that promote the AT2R's protective effects and reduce the consequences of cardiovascular disease, including atherosclerosis, MI and heart failure. The vistas that have opened up through the study of the AT2R hold out the hope of new and better treatments.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Faria-Costa G, Leite-Moreira A, Henriques-Coelho T. Efeitos cardiovasculares do receptor tipo 2 da angiotensina. Rev Port Cardiol. 2014;33:439–449.
