


















A importância dos canais de sódio para a normal atividade elétrica do coração é enfatizada pelo facto de as mutações (hereditárias ou de novo) nos genes que codificam esses canais ou as proteínas a esses associadas provocarem síndromes arritmogénicas como a síndrome de Brugada e a síndrome do QT longo. O objetivo deste trabalho é proceder a uma revisão bibliográfica sobre as mutações no complexo dos canais de sódio responsáveis por doença cardíaca e as implicações da relação estrita entre a genética e a clínica das principais canalopatias cardíacas, nomeadamente no nível do diagnóstico, da estratificação do risco, do prognóstico, do rastreio de parentes e terapêutica.
MétodosFoi usada a base de dados online Pubmed® para pesquisar os artigos publicados nessa área, em revistas indexadas. Recorreu‐se à MeSH Database para definir a seguinte query: “Mutation [Mesh] AND Sodium Channels [Mesh] AND Heart Diseases [Mesh]” e incluíram‐se artigos publicados nos últimos 15 anos, escritos em inglês ou português e referentes à investigação em humanos.
ConclusõesNos últimos anos, grandes avanços foram feitos no esclarecimento da base genética e molecular dessas síndromes. A maior compreensão dos mecanismos fisiopatológicos subjacentes demonstrou a importância da relação entre o genótipo e o fenótipo e permitiu efetuar progressos na abordagem clínica desses pacientes. Todavia, é ainda necessário melhorar a capacidade de diagnóstico, aprimorar a estratificação do risco e desenvolver novas terapêuticas específicas de acordo com o binómio genótipo‐fenótipo.
The importance of sodium channels for the normal electrical activity of the heart is emphasized by the fact that mutations (inherited or de novo) in genes that encode for these channels or their associated proteins cause arrhythmogenic syndromes such as the Brugada syndrome and the long QT syndrome (LQTS). The aim of this study is to conduct a review of the literature on the mutations in the sodium channel complex responsible for heart disease and the implications of a close relationship between genetics and the clinical aspects of the main cardiac channelopathies, namely at the level of diagnosis, risk stratification, prognosis, screening of family members and treatment.
MethodsThe online Pubmed® database was used to search for articles published in this field in indexed journals. The MeSH database was used to define the following query: “Mutation [Mesh] AND Sodium Channels [Mesh] AND Heart Diseases [Mesh]”, and articles published in the last 15 years, written in English or Portuguese and referring to research in human beings were included.
ConclusionsIn the past few years, significant advances have been made to clarify the genetic and molecular basis of these syndromes. A greater understanding of the underlying pathophysiological mechanisms showed the importance of the relationship between genotype and phenotype and led to progress in the clinical approach to these patients. However, it is still necessary to improve diagnostic capacity, optimize risk stratification, and develop new specific treatments according to the genotype‐phenotype binomial.
As canalopatias cardíacas constituem um grupo heterogéneo de doenças cardíacas hereditárias causadas por mutações nos genes que codificam os canais iónicos expressos no coração (envolvidos nas correntes de Na+ (INa), K+ (IK) e Ca2+ (ICa)) e/ou as proteínas que regulam a sua função1–3. Essas mutações originam diferentes fenótipos de acordo com as alterações incitadas nas correntes de sódio e outros iões, conferem maior suscetibilidade para ocorrência de síncope, convulsões e arritmias, embora, na maioria das vezes, não existam defeitos cardíacos estruturais subjacentes4. Tal evidencia a importância dos canais iónicos, nomeadamente dos canais de sódio (CNa), na génese e propagação do potencial de ação (PA) e, consequentemente, na excitabilidade cardíaca2,3,5–7.
As arritmias desencadeadas são potencialmente fatais e a morte súbita cardíaca (MSC) constitui, frequentemente, a primeira manifestação dessas doenças4,8. A morte súbita (MS) é uma das causas mais comuns de morte por patologia cardiovascular e, na população adulta ocidental, as canalopatias cardíacas (1‐2%) são uma das patologias predisponentes mais frequentemente diagnosticadas, a par das cardiomiopatias (10‐15%) e da doença coronária (75%)9. Na verdade, alguns estudos mostram que as canalopatias cardíacas são responsáveis por aproximadamente 1/3 dos casos de MS em jovens com autópsia negativa e por até 50% dos casos de MSC arrítmica10,11.
As principais arritmias hereditárias causadas por disfunção dos canais iónicos são a síndrome de Brugada (SBr), a síndrome do QT longo (SQTL), a síndrome do QT curto (SQTC) e a taquicardia ventricular polimórfica catecolaminérgica (TVPC)4,12. Contudo, a sua prevalência na população em geral é difícil de estimar11,13–15. Além das patologias supracitadas, a síndrome de pré‐excitação, a fibrilhação ventricular idiopática e casos raros de cardiomiopatias familiares também se associam a mutações nos canais iónicos4,12.
Nas últimas duas décadas, o conhecimento sobre os mecanismos genéticos e moleculares subjacentes às arritmias (mormente as de cariz hereditário – Tabela 1), foi vastamente alargado e diversas mutações e/ou variantes genéticas têm sido descritas16,17.
Principais genes associados às arritmias hereditárias
| Patologia | Genes (% de envolvimento) | Prevalência |
|---|---|---|
| Arritmias hereditárias na ausência de anomalias cardíacas estruturais | ||
| Síndrome de Brugada | SCN5A (20‐30%) | 1:3.300 a 1:10.000* |
| Síndrome do QT longo | KCNQ1 (30‐35%), KCNH2 (25‐30%), SCN5A (5‐10%) | 1:2.500* |
| Taquicardia ventricular polimórfica catecolaminérgica | RYR2 (60‐65%), CASQ2 (<5%) | 1:10.000* |
| Doença cardíaca da condução | SCN5A (5%) | |
| Síndrome do QT curto | Nenhum gene dos 3 conhecidos representa>5% da doença | |
| Fibrilhação auricular | Nenhum gene dos conhecidos representa>5% da doença | |
| Arritmias hereditárias na presença de anomalias cardíacas estruturais | ||
| Cardiomiopatia arritmogénica ventricular direita/displasia ventricular direita arritmogénica | PKP2 (25‐40%), DSG2 (5‐10%), DSP (2‐12%), DSC2 (2‐7%) | |
| Cardiomiopatia dilatada | TTN (≈25%) | |
| Cardiomiopatia hipertrófica | MYBPC3 (30‐40%), MYH7 (20‐30%), TNNT2 (10%), TNNI3 (7%) | |
Apesar de várias mutações em diversos canais iónicos afetarem as correntes elétricas cardíacas, nesta revisão bibliográfica aborda‐se apenas o que concerne às correntes de sódio, designadamente: a estrutura dos CNa e o seu papel na excitabilidade cardíaca, as mutações no complexo dos CNa, os fenótipos associados e as implicações da relação entre a genética e a clínica no nível do diagnóstico, a estratificação do risco, o prognóstico e a terapêutica, fundamentalmente da SQTL e da SBr.
MétodosFoi feita uma revisão narrativa da literatura acerca do tema Canalopatias cardíacas: o papel das mutações nos canais de sódio. Para efetuar a pesquisa dos artigos publicados neárea, foi usada a base de dados online Pubmed® e recorreu‐se à MeSH Database para selecionar os termos MeSH e definir a seguinte query: “Mutation [Mesh] AND Sodium Channels [Mesh] AND Heart Diseases [Mesh]”.
Com a aplicação dos critérios de inclusão preestabelecidos, foram incluídos apenas artigos publicados nos últimos 15 anos, escritos em inglês ou português e referentes à investigação em humanos. Adicionalmente, foi tido em conta o factor de impacto.
Incluíram‐se ainda outras referências bibliográficas, algumas das quais com data de publicação anterior a 2002, com vistas a aprofundar conteúdos relevantes citados nos artigos inicialmente pesquisados.
Estrutura e função dos canais de sódioOs canais de sódio (CNa) são proteínas transmembranares compostas por uma subunidade α em conjunto com uma ou duas subunidades β (Figura 1)2. Existem vários tipos de subunidades α, diferentemente expressas consoante o tipo de tecido e codificadas por uma família de 10 genes distintos (Tabela 2)18,19. A principal subunidade α expressa no coração denomina‐se Nav1.5 (tal como o canal de sódio em que se integra) e é codificada pelo gene SCN5A (sodium channel, voltage gated, type V alpha subunit), esse último constituído por 28 exões e que abrange mais de 100kb no cromossoma 3p2218,20.
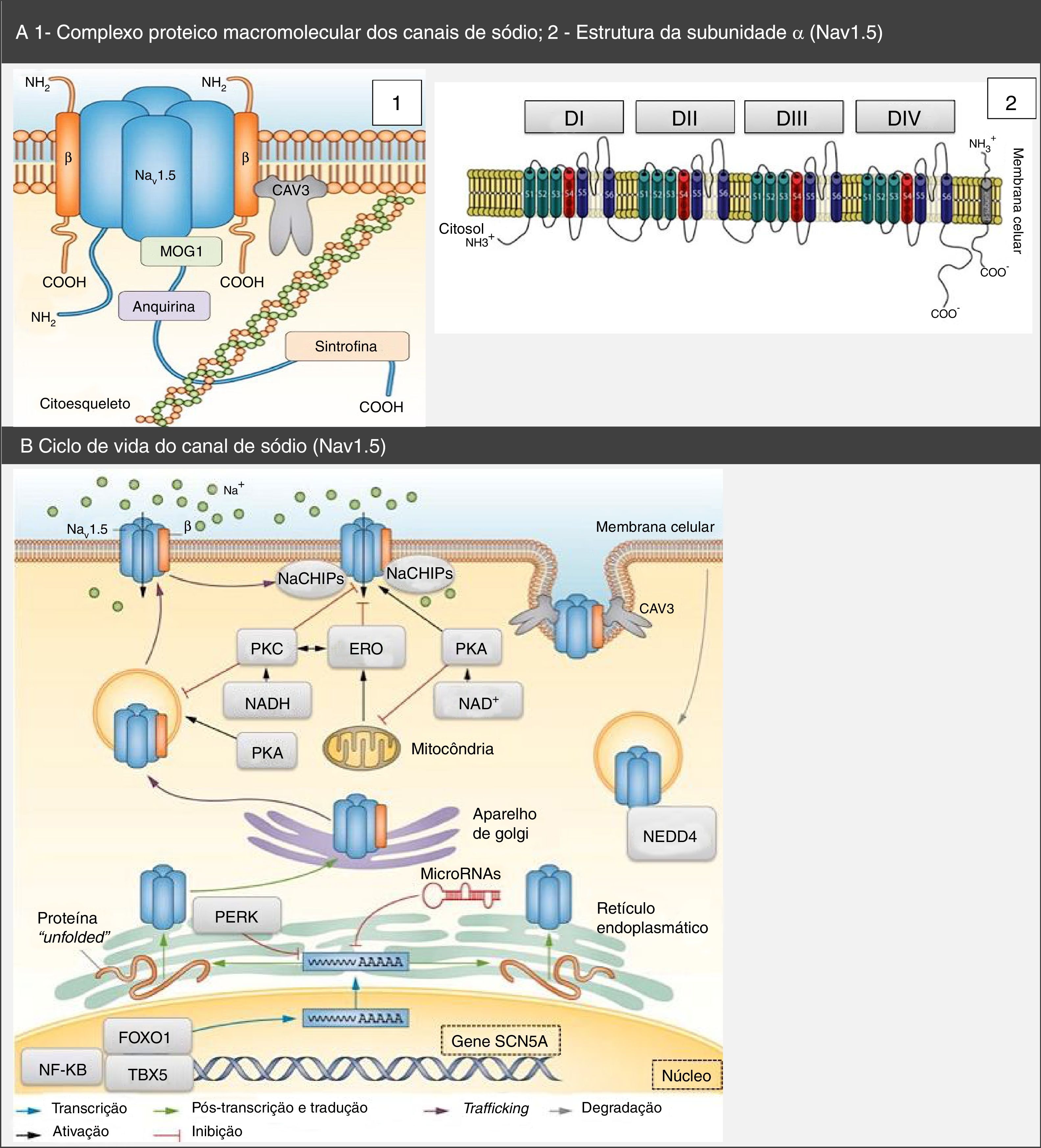
Complexo proteico macromolecular, subunidades α e ciclo de vida dos canais de sódio Nav1.5. (A) O canal Nav1.5 integra um complexo macromolecular e interatua com diversas proteínas, entre as quais: subunidades β, caveolina‐3, MOG1, anquirina, sintrofina e citoesqueleto. Retirado e adaptado de Liu et al. (2014)7 e de Amin et al. (2010).22(B) O ciclo de vida do Nav1.5 inicia‐se no núcleo, onde ocorre a transcrição do gene SCN5A e respetiva regulação por fatores de transcrição (FOXO1, NF‐KB e TBX5). Contudo, os microRNAs também regulam os níveis de mRNA. No retículo endoplasmático ocorre a tradução proteica e, após ocorrer folding apropriado e assembly de proteínas, essas são transportadas para a membrana celular (trafficking). Mutações ou variantes de splicing podem levar à formação de uma proteína Nav1.5 misfolded e pode ser ativada a via PERK com vista ao down regulation dos seus níveis de mRNA. A PKA, PKC, o stresse oxidativo (ERO) e os estados metabólicos (NADH e NAD+) podem modular o trafficking do canal. O NEDD4 regula a degradação mediada pela ubiquitina. Retirado e adaptado de Liu et al. (2014)7.
CAV3: caveolina‐3; ERO: espécies reativas de oxigénio; FOXO1: forkhead box protein O1; MOG1: Ran guanine nucleotide release factor; NaChIP: Na+‐channel‐interacting protein; NEDD4: E3 ubiquitin‐protein ligase NEDD4; NF‐κB: factor nuclear NF‐κB; PERK: factor de iniciação da tradução eucariótica 2α‐cinase 3; PKA: proteína cínase dependente de AMPc (proteína cínase A); PKC: proteína cínase C; TBX5: fator de transcrição T‐box TBX5.
Subunidades α dos canais de sódio
| Proteína | Tecido com expressão major | Gene | Cromossoma |
|---|---|---|---|
| Nav1.1 | SNC e SNP | SCN1A | 2q24 |
| Nav1.2 | SNC e SNP | SCN2A | 2q23‐24 |
| Nav1.3 | SNC e SNP | SCN3A | 2q24 |
| Nav1.4 | Musculoesquelético | SCN4A | 17q23‐25 |
| Nav1.5 | Coração | SCN5A | 3p21 |
| Nav1.6 | SNC e SNP | SCN8A | 12q13 |
| Nav1.7 | SNP | SCN9A | 2q24 |
| Nav1.8 | SNP | SCN10A | 3p21‐24 |
| Nav1.9 | SNP | SCN11A | 3p21‐24 |
| Nav2.1 (Nax) | Células Glia | SCN6/7A | 2q21‐23 |
SNC: sistema nervoso central; SNP: sistema nervoso periférico.
Retirado e adaptado de England e Groot (2009).19
A regulação da transcrição do gene SCN5A é influenciada por vários fatores, entre eles: existência de três promotores, fatores de transcrição e microRNAs com ação pós‐transcrição. Já foram descritas mais de 10 isoformas resultantes do splicing desse gene, a isoforma mais abundante no coração humano é a SCN5A‐003 (isoforma do adulto)7,18,20.
A subunidade α tem 227 kDa e consiste numa proteína transmembranar com quatro domínios homólogos (DI‐DIV) conetados por ansas citoplasmáticas, cada um dos quais com seis segmentos transmembranares em α‐hélice (S1‐6) conetados por ansas intra e extracelulares. Apresenta ainda um terminal C (carboxilo) e um terminal N (amino), ambos citoplasmáticos5,6,21.
O poro central é formado pelos quatro segmentos S5 e S6da subunidade α, nomeadamente, pelas ansas extracelulares que os conectam. Apresenta permeabilidade seletiva para o sódio, que se desloca através desse de acordo com o gradiente eletroquímico. Os segmentos S1 a S4 funcionam como sensores de voltagem. Contudo, o último apresenta a particularidade de ter carga positiva2,5,18,21.
À semelhança dos restantes canais dependentes de voltagem, os CNa exibem alterações da sua conformação, um processo denominado gating e que permite definir os três estados funcionais do canal (aberto, inativo ou fechado), de acordo com o potencial de membrana. Essas alterações ocorrem na subunidade α, principal responsável pela regulação da despolarização da membrana das células excitáveis2,18,22.
As subunidades β são proteínas de aproximadamente 30‐40 kDa, com um único segmento transmembranar, um terminal C intracelular e um terminal N extracelular5. Essas subunidades associam‐se à subunidade α do CNa (Figura 1) e, assim, não só modulam a sua expressão na superfície celular e o processo de gating como também permitem a ligação ao citoesqueleto e a outras proteínas de interação. Efetivamente, as subunidades β são capazes de aumentar o tráfego dos canais para a membrana, com consequente aumento da INa5,18.
Existem quatro tipos de subunidades β (β1, β2, β3 e β4), codificadas pelos genes SCN1B, SCN2B, SCN3B e SCN4B, respetivamente. Essas associam‐se preferencialmente a diferentes subunidades α, consoante o tipo de tecido no qual se expressam5,18,20,23.
Para além das subunidades β, existem outras proteínas com capacidade para interferir e modular a função do Nav1.5 (anquirina‐G, calmodulina, caveolina‐3, sintrofina α1, placofilina‐2, Ran guanine nucleotide release factor (MOG1), desidrogénase do glicerol‐3‐fosfato 1‐like (GPD1L), factor homólogo 1B do Fibroblast Growth Factor (FHFGF‐1B), lígases da ubiquitina Nedd4‐like, entre outras) que integram um complexo macromolecular (Figura 1)5,6,18,20,21,24,25.
O papel dos canais de sódio na excitabilidade cardíacaO potencial de ação cardíaco é gerado por correntes iónicas despolarizantes (INa; ICa) e repolarizantes (IK)22. Os CNa têm um papel determinante na iniciação do PA através da génese da INa, são expressos na membrana dos cardiomiócitos auriculares e ventriculares e no tecido de condução especializada21–23. Contudo, embora a sua expressão seja abundante no feixe de His, nos seus ramos e nas fibras de Purkinje, a sua expressão é baixa ou ausente nos nós sinusal e auriculoventricular6,21.
No miocárdio ventricular, durante a diástole, o potencial elétrico transmembranar (de repouso) é aproximadamente ‐85mV e os CNa encontram‐se fechados. Quando um estímulo despolariza a membrana, os segmentos S4 dos quatro domínios movimentam‐se simultaneamente para o exterior, o canal abre e há passagem de Na+ para o meio intracelular de acordo com o gradiente eletroquímico5,6,18,22. Assim, gera‐se a INa, principal responsável pela fase de despolarização rápida do PA (fase 0), que aumenta rapidamente até atingir o seu pico (INapico) e diminui milissegundos depois18,23.
Na inativação dos CNa, a ansa entre os domínios III e IV (porta de inativação) funciona como «tampa» e os canais fecham gradualmente em cerca de 1ms18,26. Note‐se que os CNa sofrem várias alterações conformacionais que se traduzem em diferentes estados de inativação (inativação rápida, intermédia e lenta), que, por sua vez, têm diferentes tempos de recuperação18,22. Porém, no fim da fase 0 a maioria (≈99%) dos CNa está inativada, o que impede a passagem de iões. Assim permanecem até à repolarização da membrana celular, momento em que se recuperam da inativação e ficam novamente disponíveis para ser ativados durante a fase 45,18.
Contudo, durante a fase 2 do PA, uma pequena fração dos CNa (< 1% do total de CNa disponíveis) pode manter condutibilidade para o Na+ e reabrir, persistindo uma pequena INa denominada late current (INatardia)5,18,27. Para além disso, alguns canais podem reativar durante a fase de repolarização (fase 3), quando a inativação ainda não está completa mas o PA permite a sua reativação, o que gera uma corrente denominada window current18,22. Essa última corresponde a menos de 1% do pico da corrente de sódio22. A essas duas correntes tem sido atribuído um papel importante na arritmogénese ventricular presente em certas patologias cardíacas, algumas delas abordadas nesta revisão5,18,22,27.
Em condições fisiológicas, os processos de ativação e inativação dos CNa são estritamente regulados de forma a assegurar a normal atividade elétrica cardíaca. As anomalias dos CNa provocam alterações importantes na eletrofisiologia cardíaca, potenciam a arritmogénese e podem resultar de alterações das propriedades de gating ou da cinética da INa. Essas alterações modificam a disponibilidade dos canais e a amplitude da INapico ou impedem a inativação adequada dos canais, com manutenção de uma INa persistente durante o plateau do PA. Assim, a importância dos CNa na excitabilidade cardíaca é enfatizada pela ocorrência de arritmias potencialmente fatais (por exemplo: taquicardia ventricular e fibrilhação ventricular) na presença de disfunção de causa hereditária ou adquirida desses canais18,21,22.
Mutações nos canais de sódioMutações na subunidade αNas últimas décadas, o conhecimento acerca da função do gene SNC5A no nível molecular e eletrofisiológico foi amplamente alargado e vários estudos genéticos mostram que mutações nesse gene estão associadas a várias doenças cardíacas, nomeadamente arritmias cardíacas hereditárias1,5,16,18,21,22,28. Na sua maioria, as patologias associadas a mutações nos CNa são causadas por mutações que alteram a permeabilidade do canal ou o processo de gating6,21.
As mutações no gene SCN5A que levam à disfunção do CNa Nav1.5 podem ser de ganho de função, perda de função ou ambas7,18.
As mutações de perda de função resultam na diminuição da INa e associam‐se a SBr, doença do nó sinusal (DNS), fibrilhação auricular (FA), doença de Lev‐Lenégre e cardiomiopatia dilatada (CMD) (Figura 2)7,18. O mecanismo mais frequentemente envolvido é a diminuição da INapico (Figura 3)7,18,23.
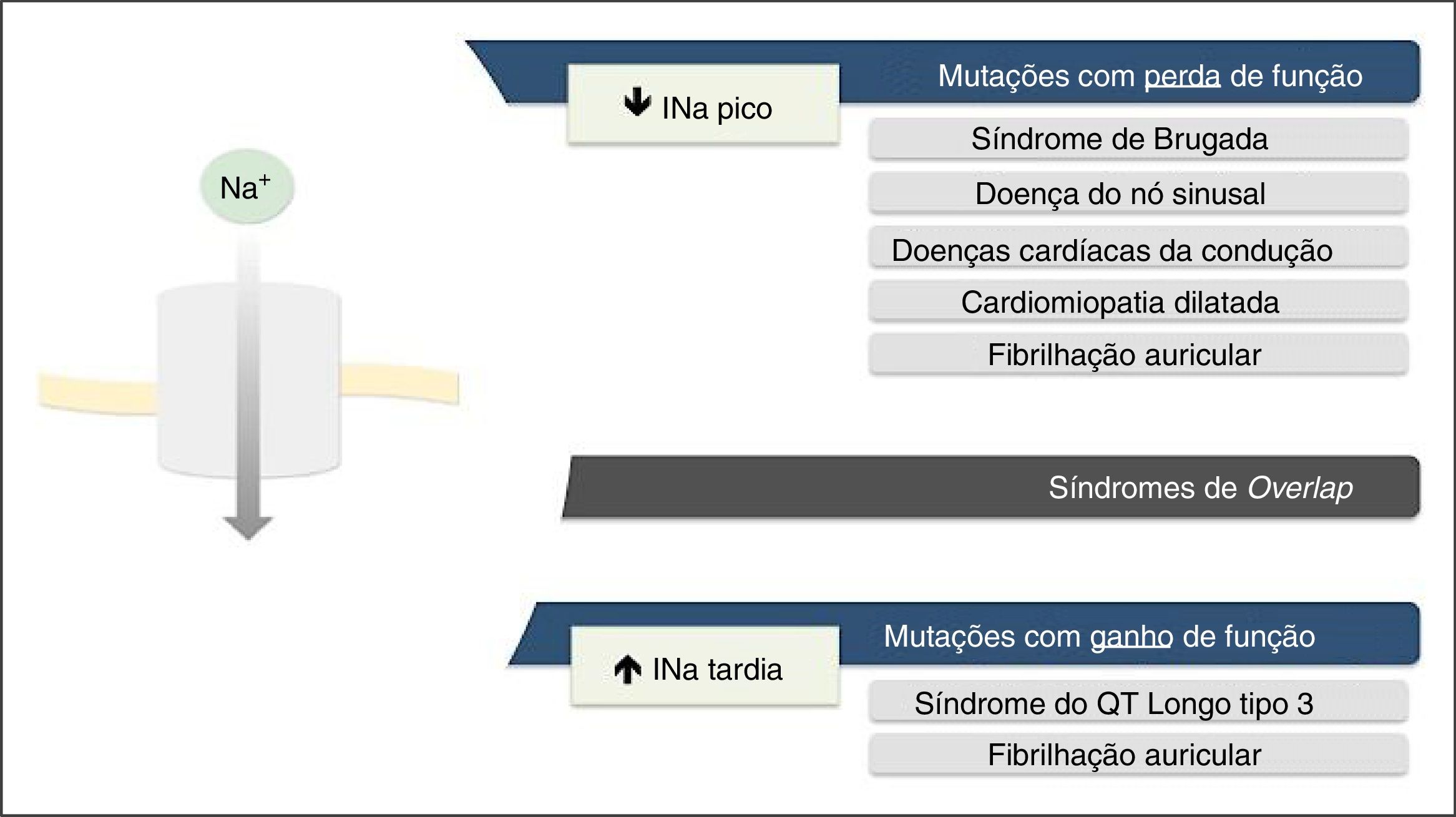
Fenótipos clínicos associados a mutações nos canais de sódio Nav1.5.
Retirado e adaptado de Liu et al. (2014)7.
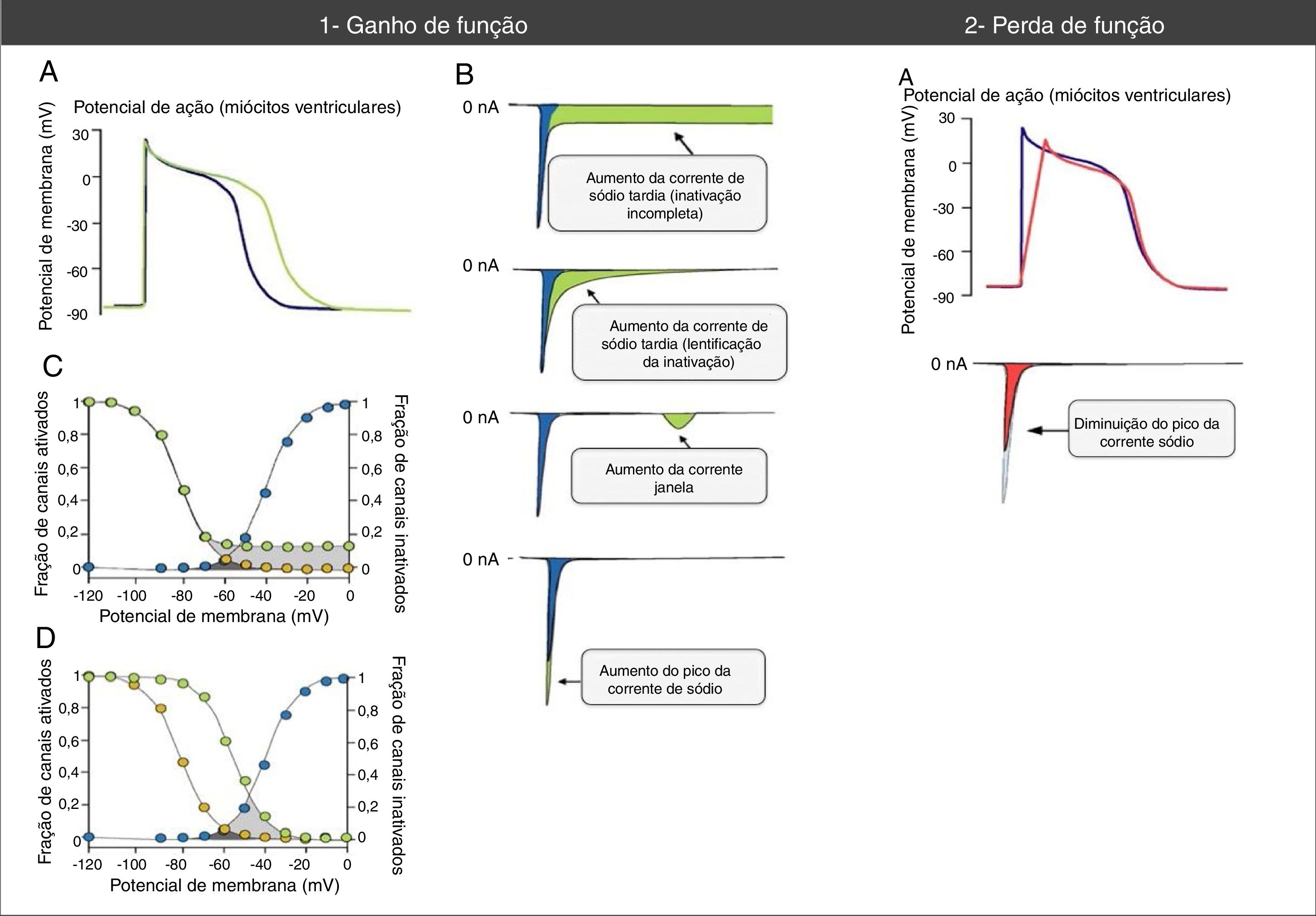
Alterações do potencial de ação e das correntes de sódio associadas a mutações nos canais Nav1.5 de ganho e perda de função. (1A) As mutações de ganho de função associam‐se a um aumento a duração do potencial de ação e podem desencadear eventos arrítmicos. (1B) Vários mecanismos podem associar‐se a ganho de função na corrente de sódio. O mecanismo mais comum é o aumento da corrente de sódio tardia (aumento anormalmente sustentado da INa durante a fase 2 do PA com prolongamento da despolarização da membrana e atraso da repolarização) que pode dever‐se a inativação incompleta ou lentificada. Outros mecanismos menos comuns são o aumento da corrente janela e o aumento da INa pico (aumento do influxo de Na+ na fase 0 do PA). (1C) O mecanismo de aumento da corrente de sódio mais frequentemente resulta da inativação incompleta dos canais de sódio (círculos verdes). (1D) No mecanismo de aumento da corrente da janela (círculos verdes), a inativação ocorre em PA mais positivos, «atrasa‐se» e alarga‐se a amplitude de voltagens durante as quais os CNa podem reativar. (2A) Nas mutações de perda de função a diminuição do pico da corrente de sódio diminui a velocidade de upstroke da fase 0 do potencial de ação e retarda a condução elétrica no coração.
Retirado e adaptado de Amin et al. (2010).22
As mutações de ganho de função resultam num aumento da INa e associam‐se a SQTL3 (Figura 2). Existem também algumas mutações de ganho de função associadas à FA e à CMD7,18. O mecanismo mais frequentemente implicado consiste no aumento da INatardia7,18,23. Contudo, existem outros mecanismos, tais como: aumento da INapico, diminuição da taxa de inativação ou aumento da corrente de janela (Figura 3)18,22.
Raramente, as mutações podem originar simultaneamente redução da INapico e aumento de INatardia e cursam, assim, com perda e ganho de função, respetivamente7,23.
Mutações nas subunidades βAs mutações na subunidade β1 foram identificadas em pacientes com SBr, FA e doença cardíaca da condução (DCC) (Tabela 3). Pensa‐se que o mecanismo envolvido nesses fenótipos curse com diminuição da densidade da INa (perda de função). No entanto, dado o número limitado de pacientes portadores dessas mutações, não é possível esclarecer cabalmente o mecanismo envolvido ou a relação genótipo‐fenótipo20,23,25.
Mutações nas proteínas do complexo macromolecular do canal de sódio
| Gene | Proteína | Efeito normal na INa | Mutações | Efeito da mutação | Fenótipo |
|---|---|---|---|---|---|
| SCN1B | β1 | (↓) INa tardia | Trp179X | (Ø‐) Ativação, (Ø‐) ISS, (Ø↑) INa pico | SBr, DCC |
| (↑)T.Rec | E87Q | (Ø‐) Ativação, (Ø↑) INa pico | SBr, DCC | ||
| (↑) INa pico | R85H | (+) Ativação, ISS, (Ø↑) INa pico | FA familiar | ||
| D153N | (Ø↑) INa pico | FA familiar | |||
| R214Q | (Ø↑) INa pico | SBr, FA familiar | |||
| SCN2B | β2 | Estado de sialylation | R28Q | (+) Ativação, (↓) INa pico | FA familiar (↑) PR, (↑) PD.ST |
| (↑) Corrente tardia | R28W | (+) ISS, (+) Ativação, (↓) INa pico | FA familiar (↑) PR, (↑) PD.ST | ||
| SCN3B | β3 | (↑) INa, (↑)T.Rec | R6K, L10P e M161T | Misto, (↓) INa pico, (‐) SSI, (↓) T. Rec | FA familiar SBr |
| (+) ISS, (↑) P.Ref | A130V | (↓) INa pico | FA familiar | ||
| ISS, (↑) | V54G | (↓) INa pico (↓)Trafficking | FV idiopática, SMSI | ||
| P.Ref | V36M | (↓) INa pico (↑) INa tardia | SMSI | ||
| SCN4B | β4 | (↑) Velocidade do upstroke do PA | S206L | (↑) INa tardio | SMSI |
| (+) ISS | L179F | (↑) Corrente janela | SQTL10 | ||
| CAV3 | Caveolina 3 | Scaffolding (↓) INa tardia | F97C, S141R | (↑) INa tardia | SQTL9 |
| V14L,T78M e L79R | (↑) INa tardia | SMSI | |||
| GPD1L | GPD1L | (↑) INa por fosforilação | A280V | (↓) INa pico | SBr |
| E83K, I124V, R273C | (↓) INa pico | SMSI | |||
| RANGRF | MOG1 | (↑) Densidade de superfície (↑) INa pico | E83D | (↓) INa pico (↓) Trafficking | SBr |
| SNTA1 | Sintrofina α1 | Scaffolding | A390V | (↑) INa pico, (↑) INa tardia | SQTL12 |
| S287R, T372M, G460S | (↑) INa pico, (↑) INa tardia, (+) ISS | SMSI |
(‐): Shift hiperpolarizante; (+): Shift despolarizante; (↑): aumento; (↓): diminuição; DCC: doença cardíaca da condução; FA: fibrilhação auricular; FV: fibrilhação ventricular; ISS: Inativação steady‐state;Ø: falha; P.Ref: período refratário; PD.ST: segmento ST nas derivações pré‐cordiais direitas; SBr: síndrome de Brugada; SMSI: síndrome de morte súbita da infância; SQTL: síndrome do QT Longo; T.Rec: taxa de recuperação.
Retirado e adaptado de Adsit et al. (2013).23
A prevalência das variantes potencialmente patogénicas dos genes das subunidades β é semelhante à de outros genes minor envolvidos na SBr29,30. Efetivamente, apesar de nos últimos anos o conhecimento dos mecanismos subjacentes à SBr incidir sobretudo no gene SCN5A, o rastreio das quatro subunidades β pode conduzir a um aumento potencial do diagnóstico genético dessa síndrome até aproximadamente 5,4%30.
As mutações na subunidade β1 e β2 estão associadas à FA e o mecanismo é a alteração do gating e a diminuição da INa6,31. Em 2011, Olesen et al.32 descreveram mutações associadas à FA também na subunidade β3. Essas mutações reduzem a INa, aumentam a suscetibilidade para FA através de um de dois mecanismos: atraso da condução ou diminuição do período de refratariedade (promove a possibilidade de circuitos de reentrada)32. Além disso, as mutações no SCN3B também se associam à SBr (Tabela 3)8,23.
As mutações na subunidade β4 já foram descritas na SQTL10, atribuem ganho de função e o mecanismo mais provável consiste no aumento da INatardia23,25,30,33.
Mutações nas proteínas associadas aos canais de sódioOs CNa integram‐se num complexo macromolecular ao qual pertencem várias proteínas que participam na adesão celular, vias da transdução de sinal e citoesqueleto (Figura 1)6,7,11. Essas últimas encontram‐se ligadas ao CNa direta ou indiretamente e apresentam capacidade para modular a sua expressão, tráfego e função6,19,23. Assim, a disfunção dessas proteínas contribui para a fisiopatologia das canalopatias cardíacas6,22,23,28.
Mutações em várias dessas proteínas associam‐se a SQTL ou SBr (Tabela 3)5,22,34–36. A caveolina‐3 (CAV3) é uma proteína importante no tráfego de membrana e posicionamento dos canais iónicos na membrana sarcoplasmática, que regula várias correntes iónicas no coração como a INa. A sintrofina α1 (SNTA1) é uma proteína do citoesqueleto que interage com o CNa (Figura 1). As mutações com ganho de função descritas na CAV3 associam‐se a SQTL9, enquanto aquelas descritas na SNTA1 se associam a um fenótipo semelhante ao da SQTL36,15,22,28,37. Já as mutações na anquirina‐B, cuja função é a ligação de proteínas de membrana às estruturas do citoesqueleto (Figura 1), associam‐se a SQTL4, FA, entre outros15.
As mutações no gene GPD1L, que codifica a desidrogenase 1‐like do glicerol‐3‐fostato, ou no gene MOG1, que codifica uma molécula que afeta o tráfego de proteínas, já foram descritas na SBr6,15,23,25. Adicionalmente, mutações na placofilina‐2, uma proteína do desmossoma, podem dar origem à diminuição da INa e, assim, a um fenótipo similar ao do SBr15,38,39.
Fenótipos «cardíacos» associados à disfunção dos canais de sódio e proteínas interatuantesNa Tabela 4 descrevem‐se os diversos fenótipos «cardíacos» associados a mutações nos genes que codificam os CNa e as proteínas que integram o seu complexo macromolecular. As canalopatias cardíacas mais prevalentes são a SQTL (1:2500) e a SBr (1:3.300 a 1:10.000) e associam‐se, em parte, à disfunção dos CNa12. Assim, seguidamente serão abordadas apenas essas duas entidades.
Fenótipos «cardíacos» associados a disfunção dos canais de sódio e proteínas relacionadas
| Gene | Proteína | Alterações na INa | Fenótipo «cardíaco» |
|---|---|---|---|
| Canal de sódio | |||
| SCN5A | Nav1.5 | (↓) INa por diferentes mecanismos | SBr tipo 1 |
| (↑) INa tardia | SQTL tipo 3 | ||
| (↓) INa por diferentes mecanismos | DCC | ||
| (↓) INa por diferentes mecanismos | Doença de Lev‐Lenégre | ||
| (↓) INa por diferentes mecanismos | Bloqueio AV congénito | ||
| (↓) INa | DNS | ||
| (↓) INa | Atrial standstill | ||
| Fenótipos moleculares diferentes e discordantes | FA | ||
| Fenótipos moleculares diferentes e discordantes | CMD | ||
| (↑) INa tardia / (↓) INa | SMSI | ||
| Combinação de fenótipos moleculares presentes noutras entidades clínicas | Síndrome de Overlap | ||
| Proteínas do complexo macromolecular do canal de sódio | |||
| SCN1B | Subunidade β1 | (↓) INapico | SBr tipo 5 |
| (↓) INapico | DCC | ||
| (↓) INapico | FA | ||
| SCN2B | Subunidade β2 | (↓) INapico | FA |
| SCN3B | Subunidade β3 | (↓) INapico | SBr tipo 7 |
| (↓) INapico | FA | ||
| (↓) INapico | DCC | ||
| (↓) INa pico / (↑) INa tardia | SMSI | ||
| (↓) INapico | FV idiopática | ||
| SCN4B | Subunidade β4 | (↑) INa tardia | SQTL tipo 10 |
| (↑) INa tardia | SMSI | ||
| SNTA | Sintrofina α1 | (↑) INa tardia / (↑) INapico | SQTL tipo 12 |
| (↑) INa tardia / (↑) INapico | SMSI | ||
| RANGRF | MOG1 | (↓) INapico | SBr tipo 8 |
| CAV3 | Caveolina‐3 | (↑) INa tardia | SQTL tipo 9 |
| (↑) INa tardia | SMSI | ||
| GPD1L | Desidrogenase 1‐like do glicerol‐3‐fosfato | (↓) INapico | SBr tipo 2 |
| (↓) INapico | SMSI | ||
| PTPH1 | Tirosina fosfátase H1 | ‐ | |
| NEDD4L | Nedd4‐2/Nedd4‐like | ‐ | |
| CALM | Calmodulina | ‐ | |
| CAMK2D | Proteína cínase 2 delta dependente do cálcio/calmodulina | ‐ | |
| SAP97 | SAP97 | ‐ | |
| YWHAH | 14‐3‐3‐a | ‐ | |
| FGF13 | FGF13 | ‐ | |
| ANK3 | Anquirina‐G | ‐ | |
| ACTN2 | Actinina α2 | ‐ | |
| PKP2 | Placofilina‐2 | Cardiomiopatia arritmogénica | |
| DSG2 | Desmogleína‐2 | Cardiomiopatia arritmogénica | |
| TCAP | Teletonina | ‐ | |
| ZASP | Banda Z | ‐ | |
AV: auriculoventricular; CMD: cardiomiopatia dilatada; DCC: doença cardíaca da condução; DNS: doença do nó sinusal; FA: fibrilhação auricular; SBr: síndrome de Brugada; SMSI: síndrome da morte súbita da infância; SQTL: síndrome de QT longo.
Retirado e adaptado de Wilde e Brugada (2011),5 Remme (2013),6 Abriel (2010)15 e Adsit et al. (2013).23
A síndrome de Brugada (SBr) foi descrita pela primeira vez em 1992 como uma síndrome pautada por um padrão eletrocardiográfico típico, ausência de anomalias cardíacas estruturais e história familiar de morte súbita. Desde então, progressos foram feitos na compreensão da sua fisiopatologia e na identificação da sua base genética1,40,41.
A SBr é uma síndrome rara, hereditária, com uma prevalência estimada de 1/3.300 a 1/10. 000, para a qual já foram descritas diferenças étnicas e geográficas12,14,42. Afeta adultos relativamente jovens (< 40 anos), mais frequentemente do sexo masculino, com história familiar de morte súbita em 20‐50% dos casos21,43,44. Aliás, estima‐se que a SBr seja responsável por pelo menos 4% de todos os casos de morte súbita e pelo menos 20% dos casos de morte súbita em indivíduos sem alterações cardíacas estruturais8,45.
A ausência de anomalias cardíacas estruturais era, classicamente, uma característica da SBr41. No entanto, anomalias estruturais ligeiras nos ventrículos direito e esquerdo têm sido descritas em vários estudos46.
A apresentação clínica é muito variável. A maioria dos indivíduos encontra‐se assintomática quando do diagnóstico e esse último é feito na sequência de um ECG de rotina em 58% dos casos ou ainda na sequência de rastreio familiar em 37% dos casos44. Porém, a MSC pode ser a primeira manifestação da doença, uma vez que aqueles indivíduos apresentam risco aumentado para desenvolver taquiarritmias, nomeadamente taquicardia ventricular polimórfica (TVP) e fibrilhação ventricular (FV)22,47.
Estima‐se que a taxa de eventos arrítmicos por ano em indivíduos assintomáticos seja de cerca de 0,5%, ocorrendo mais frequentemente em repouso e durante o sono, mas também na presença de febre ou após refeições abundantes1,44,48. De facto, a febre é um dos fatores que podem provocar ou exacerbar o padrão eletrocardiográfico da SBr e despoleta arritmias potencialmente fatais em 27% dos casos21,49.
O diagnóstico faz‐se através de critérios clínicos, presença de padrão típico de alterações eletrocardiográficas e exclusão de outras etiologias que podem mimetizar a SBr, nomeadamente por desencadear elevação do segmento ST (Figura 4)45,50,51. Testes de provocação com fármacos bloqueadores dos CNa (Tabela 5) podem ser feitos para provocar as alterações eletrocardiográficas da SBr (Figura 5), o que possibilita o diagnóstico naqueles indivíduos com padrão eletrocardiográfico transitório14,41,45,52. Nesses casos, pode ser também usado o Holter, que, através de uma monitoração prolongada, pode permitir fazer o diagnóstico de alterações intermitentes.1
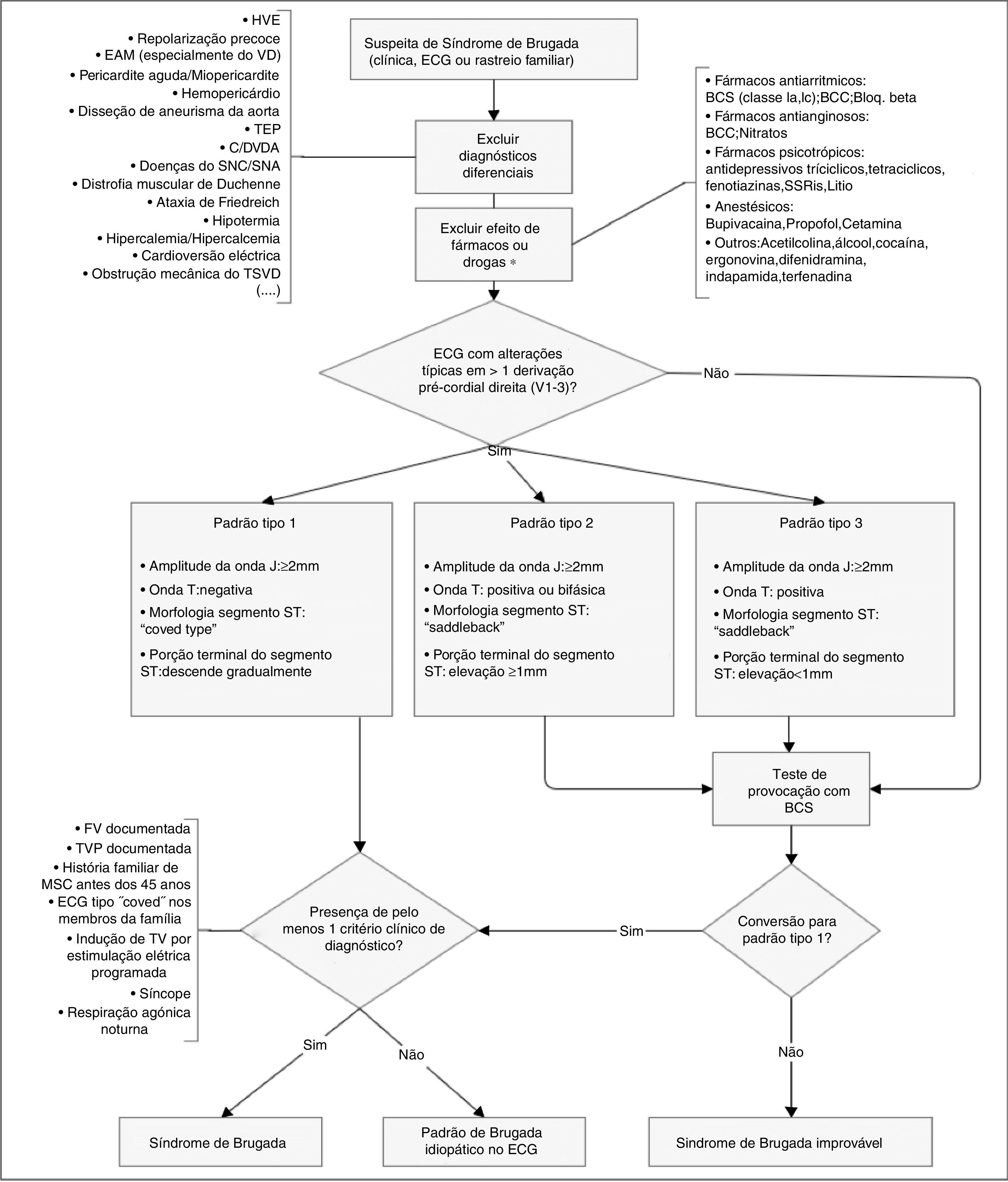
Algoritmo diagnóstico de síndrome de Brugada. Existem três padrões de alterações eletrocardiográficas nas derivações pré‐cordiais direitas (V1‐V3). O tipo 1 é considerado diagnóstico, contrariamente aos tipos 2 e 3 (na presença dos quais se devem fazer testes de provocação com BCS). Outras alterações eletrocardiográficas que podem estar presentes no SBr são: prolongamento do intervalo PR e bloqueio de ramo direito. O diagnóstico definitivo faz‐se na presença de elevação do segmento ST do tipo 1 em pelo menos uma derivação de V1‐V3 e quando se verifica um dos critérios clínicos apresentados na figura. BCC: bloqueadores dos canais de cálcio; BCS: bloqueadores dos canais de sódio; Bloq.beta: bloqueadores beta; C/DVDA: cardiomiopatia/displasia ventricular direita arritmogénica; EAM: enfarte agudo do miocárdio; ECG: eletrocardiograma; FV: fibrilhação ventricular; HVE: hipertrofia ventricular esquerda; SNA: sistema nervoso autónomo; SNC: sistema nervoso central; SSRIs: inibidores seletivos da recaptação da serotonina; TEP: tromboembolismo pulmonar; TV: taquicardia ventricular; TSVD: trato de saída do ventrículo direito; VD: ventrículo direito. *Podem desmascarar susceptibilidade genética para SBr.
Retirado e adaptado de Berne e Brugada (2012).51
Fármacos usados nos testes de provocação para «desmascarar» a síndrome de Brugada
| Fármaco | Dose e duração | Via de administração |
|---|---|---|
| Ajmalina | 1mg/kg durante 5 minutos | EV |
| Flecainida | 2mg/kg durante 10 minutos | EV |
| 400mg | VO | |
| Pilsicainida | 1mg/kg durante 10 minutos | EV |
| Procainamida | 10mg/kg durante 10 minutos | EV |
EV: endovenosa; VO: via oral.
Retirado e adaptado de Antzelevitch et al. (2005).45
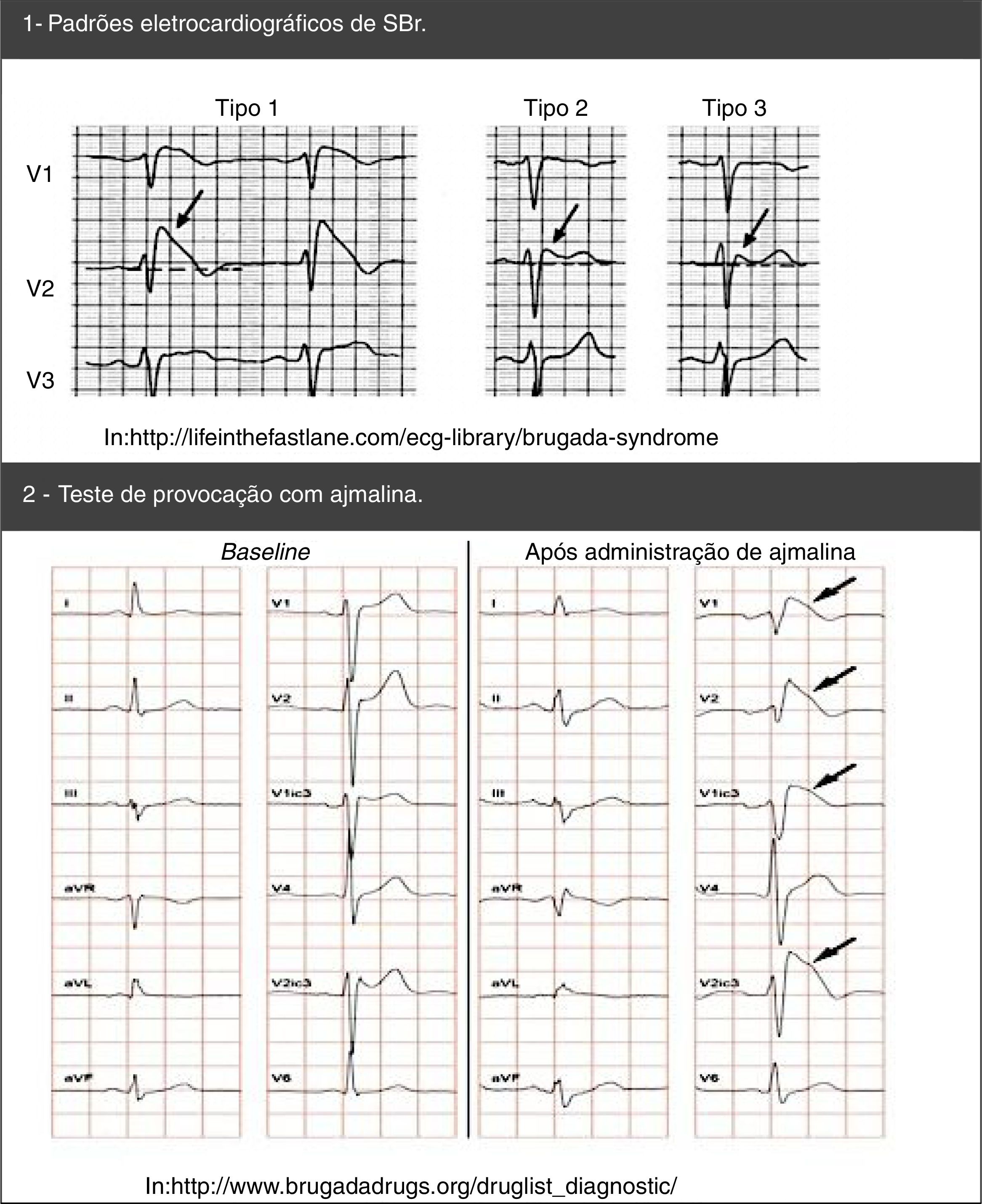
Os testes genéticos (abrangentes ou específicos para o gene SCN5A) podem ser úteis para o diagnóstico em qualquer paciente alvo de forte suspeita clínica de SBr, de acordo com a história clínica e familiar e o ECG53. Note‐se que, após a identificação de uma mutação patogénica num caso índex de SBr, está indicado um rastreio genético específico nos parentes53,54.
A SBr apresenta uma elevada complexidade genética e existem vários genes que podem estar mutados nessa síndrome, embora apenas alguns se associem a alterações das INa (Tabela 6)43,54,55. Até a data foram já descritas mais de 300 mutações que reduzem a amplitude da INa através de mecanismos diversos16,43,54,56. As mutações ocorrem mais frequentemente no gene SCN5A, localizam‐se preferencialmente nos segmentos transmembranares S1‐S4 e nos segmentos envolvidos na formação do poro (S5‐S6)16,55. Contudo, apenas uma minoria (10‐30%) da totalidade de indivíduos diagnosticados com SBr apresenta positividade para uma mutação nesse gene21,43,56–58. Outros genes (Tabela 6) estão envolvidos em menos de 5% dos casos55.
Genes mutados na síndrome de Brugada
| Fenótipo | Gene | Locus | Proteína | Efeito na função | Heredita riedade | Frequência |
|---|---|---|---|---|---|---|
| Canais de sódio e proteínas associadas | ||||||
| SBr1 | SCN5A | 3p21 | Nav1.5 | (‐) | AD | 11‐28% |
| SBr18 | SCN10A | 3p22.2 | Nav1.8 | (‐) | AD | 5.0‐16.7% |
| SBr5 | SCN1B | 19q13.12 | Subunidade β1 | (‐) | AD | 1.1% |
| SBr17 | SCN2B | 11q23.3 | Subunidade β2 | (‐) | AD | <1% |
| SBr7 | SCN3B | 11q24.1 | Subunidade β3 | (‐) | AD | <1% |
| SBr2 | GPD1L | 3p22.3 | Desidrogenase 1‐like do glicerol‐3‐fosfato | (‐) | AD | <1% |
| SBr11 | RANGRF | 17p13.1 | MOG1 | (‐) | AD | <1% |
| SBr15 | SLMAP | 3p14.3 | Proteína associada ao sarcolema | (‐) | AD | <1% |
| SBr20 | PKP2 | 12p11 | Placofilina 2 | Défice da INa# | AD | <1% |
| SBr19 | HEY2 | 6q22 | Nav1.5 | (‐) | ||
| Canais de cálcio | ||||||
| SBr3 | CACNA1C | 12p13.33 | Subunidade α1c do canal de cálcio tipo L dependente de voltagem (Cav1.2) | (‐) | AD | 6.6% |
| SBr4 | CACNB2B | 10p12.33‐p12.31 | Subunidade β2 do canal de cálcio tipo L dependente de voltagem (Cav β2) | (‐) | AD | 4.8% |
| SBr10 | CACNA2D1 | 7q21.11 | Subunidade α2/δ1 do canal de cálcio dependente de voltagem (Cavα2δ1) | (‐) | AD | 1.8% |
| SBr16 | TRPM4 | 19q13.33 | Transient receptor potential cation channel subfamily M member 4 | (‐) | AD | <1% |
| Canais de potássio | ||||||
| SBr13 | KCND3 | 1p13.2 | Canal de potássio dependente de voltagem subfamília D membro 3 | (+) | AD | <1% |
| SBr6 | KCNE3 | 11q13.4 | Canal de potássio dependente de voltagem subfamília E membro 3 | (+) | AD | <1% |
| SBr9 | KCNJ8 | 12p12.1 | Canal de potássio inward rectifier 8 sensível ao ATP | (+) | AD | 2% |
| SBr14 | HCN4 | 15q24.1 | Potassium/sodium hyperpolarization activated cyclic nucleotide gated channel 4 | (+) | AD | <1% |
| SBr12 | KCNE5 | Xq22.3 | Canal de potássio dependente de voltagem subfamília E regulatory β subunidade 5 | (+) | Ligada ao X | <1% |
| SBr8 | KCNH2 | 7q35 | Kv11.1, IKr | (+) | 1‐2% | |
| SBr21 | ABCC9 | 12p12.1 | SUR2A (subunidade 2A do recetor da sulfonilureia), IK‐ATP | (+) | 4‐5% | |
Efetivamente, apenas 30‐35% dos indivíduos com diagnóstico clínico apresentam também diagnóstico genético (genótipo positivo)8. Assim, a maioria dos indivíduos afetados (aproximadamente 65%) permanece geneticamente indeterminada (genótipo negativo) e, por esse motivo, é necessário identificar novos genes de susceptibilidade para SBr16,56,59.
Recentemente, o gene SCN10A foi identificado como gene de suscetibilidade para SBr embora a sua verdadeira prevalência permaneça por esclarecer56,60. O nível de expressão e a função do CNa Nav1.8 no coração permanecem controversos. Contudo, um estudo publicado em 2014 mostra que as variantes desse gene influenciam a duração do intervalo PR e QRS, a frequência cardíaca (FC) e, ainda, o risco de arritmias56.
Já foram descritas algumas formas recessivas com mutações homozigóticas ou heterozigóticas compostas, porém a maioria das mutações patogénicas conhecidas no gene SCN5A apresenta um padrão de transmissão autossómico dominante com penetrância variável, frequentemente incompleta18,43,45.
O mecanismo mais frequentemente implicado é a diminuição da INapico por mutações no gene SCN5A (perda de função) e consequente lentificação da condução cardíaca (Figura 3)7,18,60,61. Todavia, existem diversas hipóteses para os mecanismos fisiopatológicos da SBr que envolvem tanto alterações da despolarização como da repolarização, essas últimas não abordadas neste trabalho40,51,54.
Atualmente, o genótipo dos indivíduos com SBr não apresenta implicações relevantes para o prognóstico ou a terapêutica (Tabela 7)9. Não obstante, a sua influência no risco de arritmias e prognóstico permanece em debate54. Na verdade, os dados genéticos podem constituir uma ferramenta complementar para a estratificação do risco43,61. Mutações nonsense, que originam proteínas truncadas, têm sido associadas a pior prognóstico comparativamente a outros tipos de mutações com repercussões menos marcadas na função dos CNa40,43,59,61. Um estudo retrospetivo publicado em 2009 mostra que o fenótipo é mais grave em indivíduos com mutações associadas à redução mais significativa da INa comparativamente a indivíduos com mutações associadas a menor redução61. O mesmo se verifica quando a mutação se localiza numa região transmembranar do CNa61. Outro estudo publicado em 2013 mostra que diferentes mutações no gene SCN5A têm um impacto distinto na INa, o que reforça o papel da caracterização das mutações na avaliação do risco em parentes não afetados62. Porém, permanece pouco claro o grau em que diferentes mutações conferem risco de eventos arrítmicos ou MSC e, atualmente, a estratificação do risco faz‐se somente com parâmetros clínicos53,54.
Recomendações para o tratamento da síndrome de Brugada
| Medidas gerais de alteração do estilo de vida | |
|---|---|
| Evitar fármacos que podem induzir ou agravar a elevação do segmento ST nas DPCdt | (Classe I) |
| Evitar consumo excessivo de álcool | (Classe I) |
| Na presença de febre medicar prontamente com fármaco antipirético | (Classe Ia) |
| Estratificação do risco e tratamento específico | |||
|---|---|---|---|
| Indivíduos sintomáticosa | Indivíduos assintomáticos | ||
| MSC «abortada» | CDI (Classe I) | ECG padrão tipo 1 espontâneo | Quinidina (Classe IIb) |
| TV espontânea documentada, com ou sem síncope | CDI (Classe I) | ECG padrão tipo 1 espontâneo+TV/FV induzidas por EEF | CDI (Classe IIb) |
| Síncope+ECG padrão tipo 1 espontâneo | CDI (Classe IIa) | ||
| Tempestade elétrica/arrítmicab | Isoprenalinac(Classe IIa) | ECG padrão tipo 1 induzido por fármacos e história familiar de MSC | CDI (Classe III) |
| Quinidinad (Classe IIa) | |||
| Indivíduos que se qualificam para CDI mas apresentam uma contraindicação ou que recusam CDI e/ou apresentam história de arritmias supraventriculares que necessitam de tratamento | Quinidina (Classe IIa) | ||
| Indivíduos diagnosticados com SBr e história de tempestades elétricas/arrítmicas ou choques de repetição (apropriados) por CDI | Ablação por cateter ‐ RF (Classe IIb) | ||
CDI: cardioversor desfibrilhador implantável; DPCdt: derivações pré‐cordiais direitas; FV: fibrilhação ventricular; MSC: morte súbita cardíaca; RF: radiofrequência; SBr: síndrome de Brugada; TV: taquicardia ventricular.
O único tratamento disponível que provou prevenir a MSC em pacientes com SBr foi a implantação de um cardioversor desfibrilhador implantável (CDI)1,9,44,46. Contudo, esse procedimento acarreta um risco considerável de complicações, que ocorrem em 9% pacientes/ano e que, embora raramente ameaçadoras da vida, são psicologicamente deletérias9,54. Assim, a avaliação cuidadosa dos riscos (nomeadamente do risco de arritmias) e benefícios é um processo chave nessa decisão44,54.
Um estudo de 2003 em que foram incluídos 547 indivíduos diagnosticados com SBr, portadores de padrão eletrocardiográfico diagnóstico e sem MSC «abortada» prévia, foi feito com vista à avaliação do valor prognóstico das variáveis clínicas, eletrocardiográficas e eletrofisiológicas. Os autores verificaram que o grupo de menor risco (incidência de eventos: 0,5%) se caracteriza pela ausência de episódios de síncope, padrão eletrocardiográfico apenas despoletado por fármacos antiarrítmicos e ausência de arritmias durante a estimulação ventricular programada (EVP). Já o grupo de maior risco (incidência de eventos: 27,2%) caracteriza‐se por história prévia de episódios de síncope, ECG espontaneamente anormal e presença de arritmias induzidas por EVP, sendo que indivíduos que apresentam indutibilidade de arritmias na EVP têm um risco seis vezes superior de MSC ou FV durante os dois anos subsequentes, relativamente aos que não apresentam63.
Embora alguns sejam controversos, os fatores de risco para eventos arrítmicos são vários e entre esses os sintomas são uns dos mais importantes42,46,54,64. Com efeito, os indivíduos diagnosticados após um episódio de MSC «abortada» apresentam o risco mais elevado e em 60% desses ocorre um novo evento 10 anos após o diagnóstico54. Os indivíduos com episódios de síncope têm uma taxa de eventos arrítmicos de 1,9%/ano e a presença simultânea de padrão eletrocardiográfico tipo 1 está associada a mau prognóstico46,54. Adicionalmente, existem outros parâmetros eletrocardiográficos que se associam a pior prognóstico, como, por exemplo, a presença de fragmentação do intervalo QRS no ECG, identificada em 30‐40% dos doentes54.
Síndrome de QT longoA SQTL congénita é uma síndrome arritmogénica de origem genética/hereditária, com penetrância incompleta, cuja prevalência nos indivíduos brancos é de 1:2500, um valor muito superior ao anteriormente expectável12,13,65. Representa um grupo heterogéneo de doenças e, classicamente, é dividida em duas variantes: síndrome de Romano‐Ward e síndrome de Jervell e Lange‐Nielsen (Tabela 8)1,43,65.
Subtipos de SQTL congénita
| Nome | Gene | Proteína | Corrente | Efeito na função | Frequência |
|---|---|---|---|---|---|
| Hereditariedade autossómica dominante (Romano‐Ward) | |||||
| SQTL 1 | KCNQ1 | KV7.1 | IKs | (‐) | 40‐55% |
| SQTL 2 | KCNH2 | KV11.1 | IKr | (‐) | 30‐45% |
| SQTL 3 | SCN5A | NaV1.5 | INa | (+) | 5‐10% |
| SQTL 4 | ANKB | Anquirina B | Trocador NCX, ATPase Na+/K+ | (‐) | Raro |
| SQTL 5 | KCNE1# | MinK | IKs | (‐) | Raro |
| SQTL 6 | KCNE2 | MiRP1 | IKr | (‐) | Raro |
| SQTL 7 (SAT) | KCNJ2 | Kir2.1 | IKl | (‐) | Raro |
| SQTL 8 (ST) | CACNA1C | CaV1.2α1 | ICa,L | (+) | Raro |
| SQTL 9 | CAV3 | Caveolina‐3 | INa | (+) | Raro |
| SQTL 10 | SCN4B | Subunidadeβ4 | INa | (+) | Muito raro |
| SQTL 11 | AKAP9 | Yotiao | IKs | (‐) | Muito raro |
| SQTL 12 | SNTA1 | Sintrofina‐α1 | INa | (+) | Muito raro |
| SQTL 13 | KCNJ5 | Kir 3.4 | IK‐Ach | (‐) | Muito raro |
| SQTL 14 | CALM1 | Calmodulina 1 | Redução da afinidade para o Ca2+** | Raro | |
| SQTL 15 | CALM2 | Calmodulina 2 | Redução da afinidade para o Ca2+** | Raro | |
| Hereditariedade autossómica recessiva (Jervell e Lange‐Nielsen) | |||||
| JLN1 | KCNQ1 | KV7.1 | IKs | (‐) | Raro |
| JLN2 | KCNE1# | MinK | IKs | (‐) | Raro |
(‐): perda de função; (+): ganho de função; ICa,L: correntes de Ca2+ através dos canais de cálcio tipo L dependentes da voltagem; IK‐Ach: corrente de K+ regulada pelos recetores da acetilcolina; IKl: corrente de entrada de K+, retificadora; IKr: componente rápido (retificação interna – canais de K+estão abertos quando um potencial é negativo e fechados quando o potencial é menos negativo ou positivo) da corrente de K+delayed rectifier (IKr); IKs: componente lenta da corrente de K+delayed rectifier (IKr); INa: corrente de Na+ dependente de voltagem; NCX: trocador Na+/ Ca2+; SAT: síndrome de Andersen‐Tawil; ST: síndrome de Timothy.
As mutações no gene KCNE1 podem causar quer a síndrome de Romano‐Ward (autossómica dominante; SQTL5) quer, se em homozigotia ou heterozigotia composta, a síndrome de Jervell e Lange‐Nielsen (autossómica recessiva).
A disfunção da calmodulina pode alterar a inativação dos canais de Ca2+ tipo L dependente do Ca2+ (aumenta a corrente despolarizante durante a fase 2 do potencial de ação) mas algumas mutações da calmodulina podem também associar‐se a alteração da regulação dos canais de sódio.
Retirado e adaptado de Nakano e Shimizu (2016),34**Makita et al. (2014)36 e Mizusawa (2014).69
Em 1995 e 1996, identificaram‐se os três principais genes que conferem suscetibilidade para SQTL: KCNQ1, KCNH2 e SCN5A66–68. Esses genes constituem 75% dos casos de SQTL clinicamente definida e os restantes representam, coletivamente, apenas 5%43,69. Note‐se que a SQTL se associa a alterações das correntes de sódio unicamente nos tipos 3, 9, 10 e 1222,69.
A SQTL caracteriza‐se pelo atraso da repolarização ventricular, que se traduz eletrocardiograficamente em prolongamento do intervalo QT (Figura 6)22,65. A duração do intervalo QT é dependente da inativação dos CNa, cuja alteração pode despoletar a ocorrência de arritmias26.
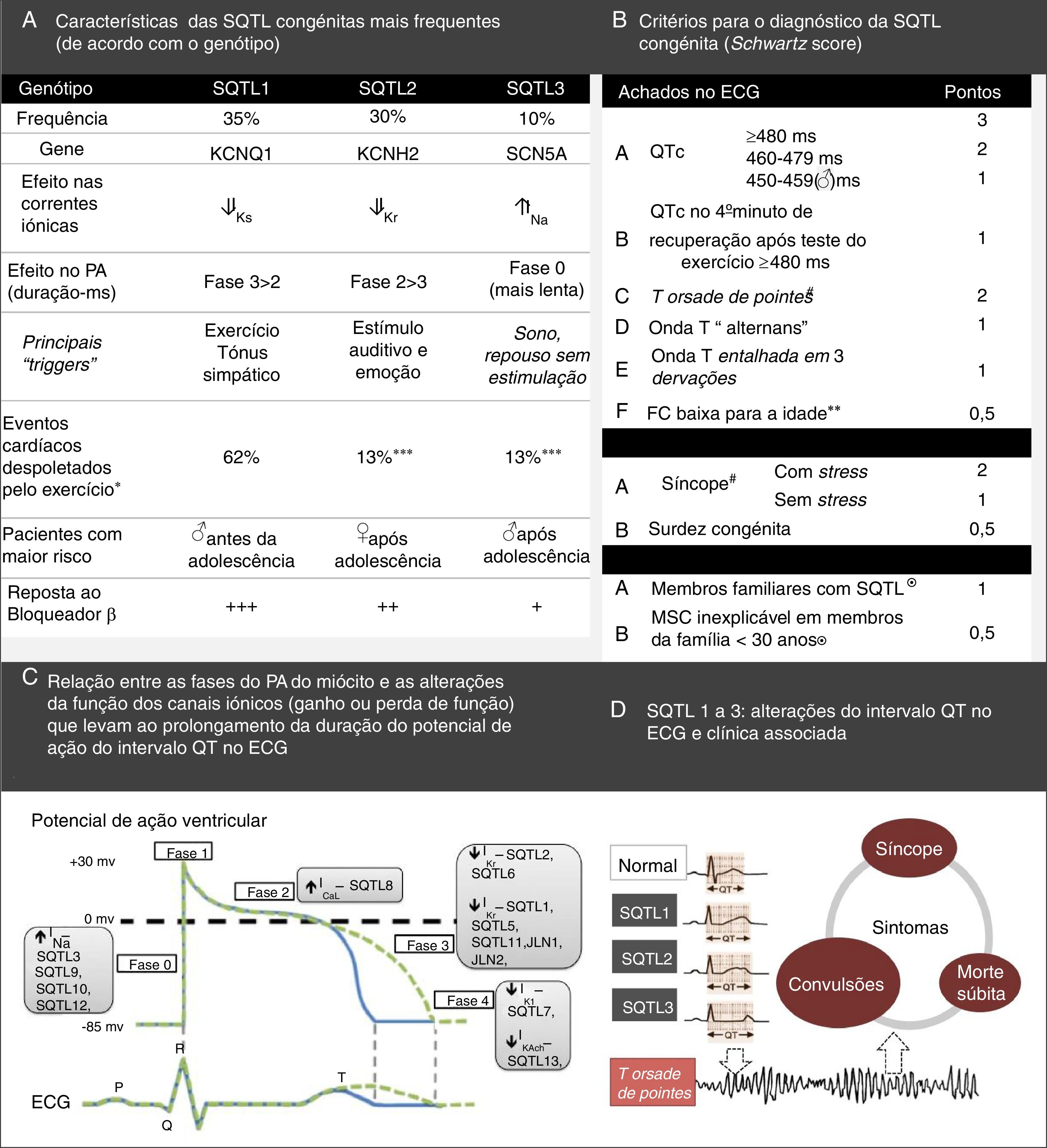
Síndrome do QT longo – principais características e critérios de diagnóstico. (B) Sistema de pontuação usado no diagnóstico da SQTL que se baseia nos achados presentes no ECG, história clínica (sintomas) e história familiar. O QTc é calculado pela fórmula de Bazett. Pontuação: ≤ 1 – baixa probabilidade de SQTL; 1,5 a 3 – probabilidade intermédia de SQTL; ≥ 3,5 – alta probabilidade de SQTL. O diagnóstico de SQTL faz‐se nos indivíduos com pontuação ≥ 3,5 nos quais não se verificam causas secundárias para o prolongamento do intervalo QT.
♂: indivíduos do sexo masculino; ♀: indivíduos do sexo feminino; FC: frequência cardíaca.
#Mutualmente exclusivos.
¿Não podem ser ambos contabilizados na mesma família.
**FC em repouso abaixo do 2° percentil para a idade.
***O baixo risco apresentado pelo exercício em pacientes SQTL2 e SQTL3 é explicado pelo facto de que ambos têm uma corrente normal de IKs, que é estimulada pela ativação do sistema nervoso simpático, que assim resulta no encurtamento da repolarização ventricular sempre que a frequência cardíaca aumenta, evita assim a probabilidade de taquiarritmias ventriculares durante o exercício.
(A) Retirado e adaptado de Furst e Aziz (2016)75 e *Schwartz et al. (2001);74(B) Retirado e adaptado de Schwartz et al. (2013);43(C) e (D) Retirado e adaptado de Giudicessi e Ackerman (2013).70
As mutações no gene SCN5A associadas à SQTL3 (ganho de função) geralmente afetam a inativação dos CNa, que fica lentificada, instável ou incompleta6,18,22,26. Consequentemente, há um aumento da INatardia com prolongamento da despolarização da membrana e atraso da repolarização18,26,65,70. Outros mecanismos possivelmente implicados são: aumento da window current, lentificação da inativação e aumento da INapico (Figura 3)6,18,22.
A primeira mutação associada à SQTL3 encontra‐se na ansa entre os domínios III e IV, correspondente à porta de inativação26. Desde então, múltiplas mutações que provocam alterações da inativação foram já identificadas e funcionalmente caracterizadas, localizando‐se em variados locais da estrutura do CNa, nomeadamente no terminal C, ao qual tem sido atribuída uma função relevante nesse processo22,26.
A SQTL congénita ocorre sobretudo em indivíduos jovens, saudáveis, sem alterações estruturais cardíacas concomitantes e associa‐se a um aumento do risco de síncope e arritmias cardíacas potencialmente fatais como a Torsade de Pointes (TdP), que degenera em FV e causa paragem cardíaca6,18,66. Na SQTL3 (diferentemente da SQTL1 e SQTL2 – Figura 6), as arritmias ocorrem usualmente em repouso, particularmente durante o sono (frequências cardíacas baixas)6,22,43,70. Note‐se que a INatardia é maior em frequências de estímulo mais lentas, o que sugere que o grau dessa corrente pode ser um determinante forte para a ocorrência de arritmias22.
O primeiro evento cardíaco (mais frequentemente síncope) ocorre usualmente em adolescentes (16±10 anos na SQTL3) e mais precocemente no sexo masculino.9,71 Porém, em 5‐10% dos casos a MSC é o evento inicial da doença e, efetivamente, a SQTL é uma das principais causas de MSC com autópsia negativa10,72.
O diagnóstico baseia‐se sobretudo na história clínica e ECG (Figuras 6 e 7)65,70. No ECG, o parâmetro mais relevante é o intervalo QT (Tabela 9), medido desde o início do complexo QRS até o fim da onda T nas derivações DII e V5 ou V665,73. Usa‐se o valor mais longo, geralmente corrigido para a FC (QTc) através da fórmula de Bazett (apesar das suas limitações para FC particularmente rápidas ou lentas)1,46,65,73.
Avaliação do intervalo QT
| 1‐ Método de correção do intervalo QT (fórmulas) | |
|---|---|
| Bazett | QT/RR1/2 |
| Fridericia | QT/RR1/3 |
| Framingham | QT+0.154 (1 – RR) |
| Hodges | QT+1.75 (FC – 60) |
| 2‐ Valores normais, borderline e prolongados do QTc calculado pela fórmula de Bazett. | |||
|---|---|---|---|
| Normal | Borderline | Prolongado | |
| 1‐15 anos | <440 ms | 440‐460 ms | >460 ms |
| Adulto (♂) | <430 ms | 430‐450 ms | >450 ms |
| Adulto (♀) | <450 ms | 450‐470 ms | >470 ms |
♂: sexo masculino;♀: sexo feminino; FC: Frequência cardíaca; RR: intervalo RR.
Retirado e adaptado de Goldenberg et al. (2006).73
Adicionalmente, é necessário excluir a presença de causas secundárias de prolongamento do intervalo QT (SQTL adquirida), como, por exemplo, fármacos, isquemia do miocárdio, cardiomiopatia, hipocalemia, hipomagnesemia, hipotermia, entre outras46,65. Uma vez excluídas, a presença num ECG de repetição de um valor de QTc ≥500ms (ou entre 480‐499ms se feito após um episódio de síncope inexplicada) é considerada diagnóstica43,46. Contudo, as SQTL tipos 1, 2 e 3 podem cursar com QTc normal no ECG em repouso em 36%, 19% e 10% dos casos, respetivamente69.
Foi criado um sistema de pontuação para diagnóstico que tem em conta vários parâmetros clínicos e eletrocardiográficos e fornece uma probabilidade de SQTL (Figura 6)43,46,70. Além disso, a monitoração com Holter e o ECG obtido durante a prova de esforço ou após infusão de adrenalina podem ser úteis em alguns casos particulares1,46,53,65,69.
Uma vez feito o diagnóstico ou mediante a ocorrência de morte súbita inexplicada num indivíduo jovem, os parentes de primeiro grau devem ser rastreados para SQTL1,65,72. Todavia, nos parentes com ECG normal não pode ser excluída SQTL43. Efetivamente, após a identificação de uma mutação patogénica num caso índex, recomenda‐se que os parentes façam um teste genético específico para a mutação em causa, com vista a identificar indivíduos portadores com intervalo QT normal43,53. Tal é importante devido ao risco de arritmias, que se estima ocorrerem em 10% dos indivíduos portadores assintomáticos1.
Além disso, recomendam‐se testes genéticos para SQTL (abrangentes ou específicos para os três principais genes) em qualquer paciente alvo de forte suspeita clínica de SQTL (baseada na história clínica, familiar e eletrocardiograma) ou, em qualquer paciente assintomático com prolongamento do intervalo QT, na ausência de outras condições clínicas que possam prolongar esse intervalo53. Efetivamente, os testes genéticos apresentam um papel relevante não só no diagnóstico da SQTL (nomeadamente, de portadores assintomáticos) e na exclusão de doença nos parentes de primeiro grau, mas também na estratificação do risco, prognóstico e tratamento (de acordo com o genótipo – Figura 7)1,43,69,70,72.
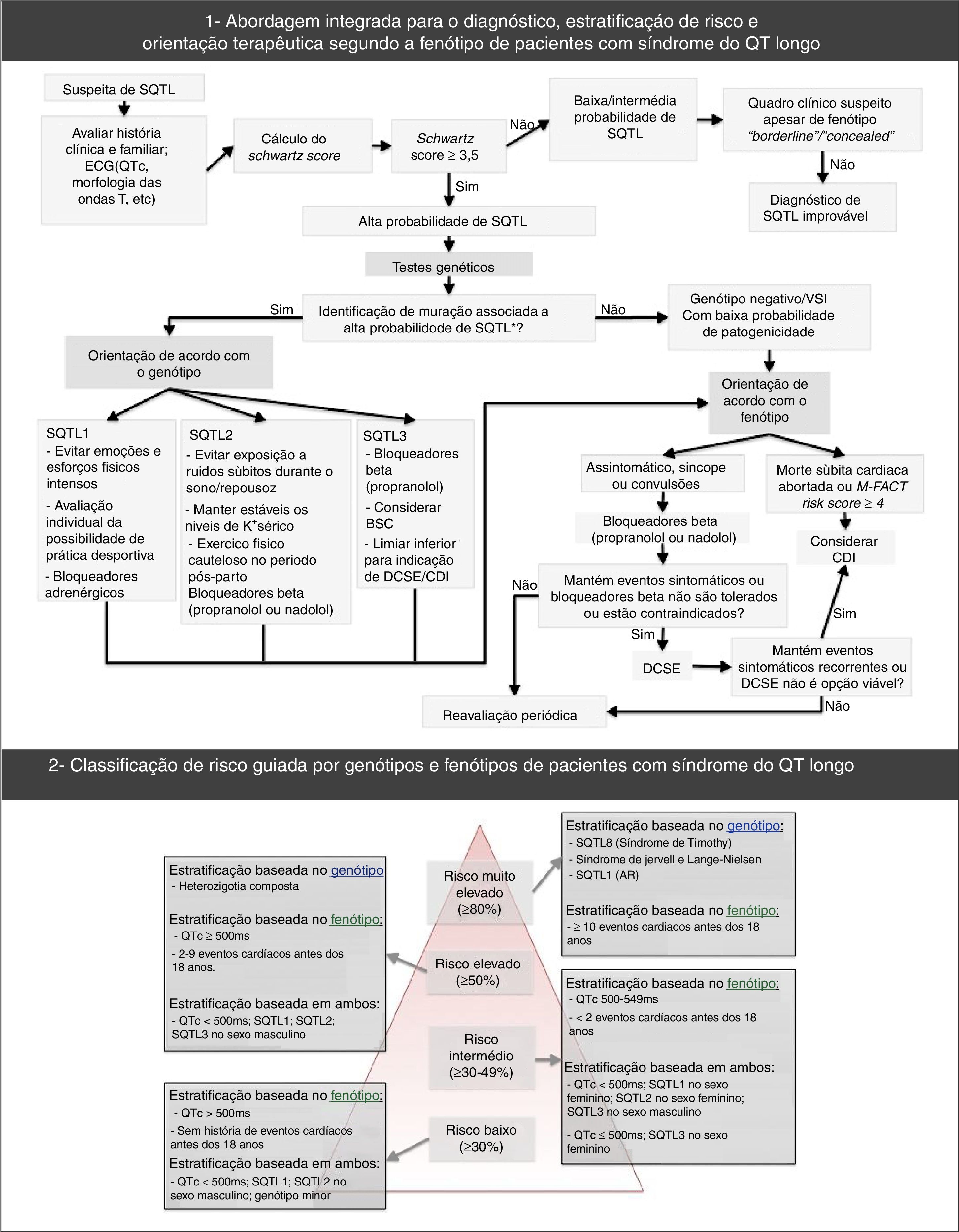
Abordagem diagnóstica, estratificação do risco e orientação terapêutica na síndrome do QT‐longo.
Retirado e adaptado de Giudessi e Ackerman (2013).70
A estratificação do risco tem em conta o fenótipo e o genótipo e faz‐se em todos os doentes, com avaliações clínicas periódicas (Figura 7)46,65,70. O risco difere de acordo com o genótipo e, adicionalmente, nos tipos genéticos mais comuns é influenciado pelo tipo e local específico das mutações, assim como pelo grau de disfunção que essas implicam43,46.
Priori et al. (2003)71 seguiram 647 indivíduos com mutações nos genes da SQTL tipos 1, 2 e 3, por um período médio de 28 anos. Os autores verificaram que 42% dos indivíduos com SQTL3 desenvolveram um primeiro evento cardíaco (ocorrência de síncope, paragem cardíaca ou MSC) antes de o indivíduo atingir os 40 anos e de iniciar a terapêutica. A incidência de paragem cardíaca ou MSC nos pacientes com SQTL3 foi de 16% e os indivíduos do sexo masculino apresentaram‐se sintomáticos mais precocemente do que aqueles do sexo feminino. Porém, dada a pequena amostra do estudo, não foi possível retirar conclusões desse achado. Adicionalmente, os autores verificaram que o intervalo QTc dos pacientes com eventos cardíacos foi significativamente mais longo do que o dos pacientes assintomáticos (subgrupo SQTL3: 523±55ms versus 481±38ms, p=0,003). Concluíram ainda que apenas um QTc superior a 498ms está associado a uma probabilidade marcadamente aumentada de eventos cardíacos. Contudo, a percentagem de indivíduos no subgrupo SQTL3 com intervalo QT normal, portadores de mutação silenciosa, foi de 10%71.
O resultado dos testes genéticos é também importante no tratamento e aconselhamento dos indivíduos afetados e parentes (Figura 7; Tabela 10)9,43. Note‐se que, por exemplo, a SQTL1 apresenta maior risco durante a atividade física comparativamente à SQTL2 e SQTL343,74,75.
Recomendações para o tratamento da síndrome QT longo
| Medidas gerais de alteração do estilo de vida | |
|---|---|
| Evitar fármacos que prolongam o intervalo QT | (Classe I) |
| Identificar e corrigir distúrbios hidroeletrolíticos | (Classe I) |
| Estratificação do risco e tratamento específico | |||
|---|---|---|---|
| Indivíduos sintomáticos | Indivíduos assintomáticos | ||
| Síncope | Bloqueadores β (Classe I) | QTc ≥ 470ms | Bloqueadores β (Classe I) |
| TV/FV documentadas | Bloqueadores β (Classe I) | QTc ≤ 470ms | Bloqueadores β (Classe IIa) |
| MSC «abortada» | CDI (Classe I) | Não tratados com bloqueadores β* | CDI (Classe III) |
| Episódios recorrentes de síncope durante terapêutica com bloqueadores β | CDI (Classe IIa) | ||
| Indivíduos com diagnóstico de SQTL que apresentam eventos durante terapêutica com bloqueadores β/ CDI | DSCE (Classe IIa) | ||
| Indivíduos de alto risco com diagnóstico de SQTL que recusam CDI ou nos quais esse está contraindicado e/ou quando os bloqueadores β não são eficazes a prevenir síncope/arritmias, não são tolerados, estão contraindicados ou são recusados | DSCE (Classe I) | ||
| Indivíduos com SQTL tipo 3 e QTc>500ms que diminui>40ms após prova oral aguda com um BCS | BSC (Classe IIa) | ||
BCS: bloqueador dos canais de sódio; CDI: cardioversor desfibrilhador implantável; DSCE: desnervação simpática cardíaca esquerda; FV: fibrilhação ventricular; MSC: morte súbita cardíaca; TV: taquicardia ventricular.
Exceto em circunstâncias especiais, o CDI não está indicado em indivíduos assintomáticos não submetidos a terapêutica com bloqueadores beta.
Adaptado de Priori et al. (2013).46
A mexiletina, a flecainida ou a ranolazina constituem opções terapêuticas «específicas» para a SQTL3 (Tabela 10), na qual os bloqueadores β podem não ser tão eficazes, pois, nesse tipo, o stress adrenérgico é um trigger com menor influência1,43,70,76. A mexiletina pode ser usada em adição aos β‐bloqueadores9,43,46,50. Porém, o seu efeito depende do tipo de mutação e pode não ser benéfica em todos os indivíduos com SQTL39,12,70.
Efetivamente, dos três principais tipos de SQTL, o tipo 3 é o que apresenta maior taxa de recorrência de arritmias em indivíduos sob tratamento com β‐bloqueadores (10‐15%)70. Tal justifica que indivíduos com SQTL3 necessitem com maior frequência de outros procedimentos mais invasivos, como desnervação simpática cardíaca esquerda e/ou implantação de CDI (Tabela 10)70.
ConclusãoAs canalopatias cardíacas são pouco frequentes na prática clínica (embora mais comuns do que se pensava), mas têm um impacto significativo na qualidade de vida e na sobrevida1,13. A sua abordagem clínica constitui um desafio devido à elevada heterogeneidade clínica e genética que as caracteriza1,76.
Apesar de inicialmente encaradas como entidades clínicas separadas com fenótipos distintos, essas síndromes podem apresentar características clínicas e genéticas que se sobrepõem (Figura 8)21. Efetivamente, além das mutações estritamente de perda ou ganho de função, existe um grande espetro de mutações associadas a anomalias diversas com diferentes repercussões na função dos CNa25. Em alguns casos, uma única mutação no gene SCN5A pode resultar em múltiplos distúrbios do ritmo e, assim, vários fenótipos podem coexistir na mesma família6,25.
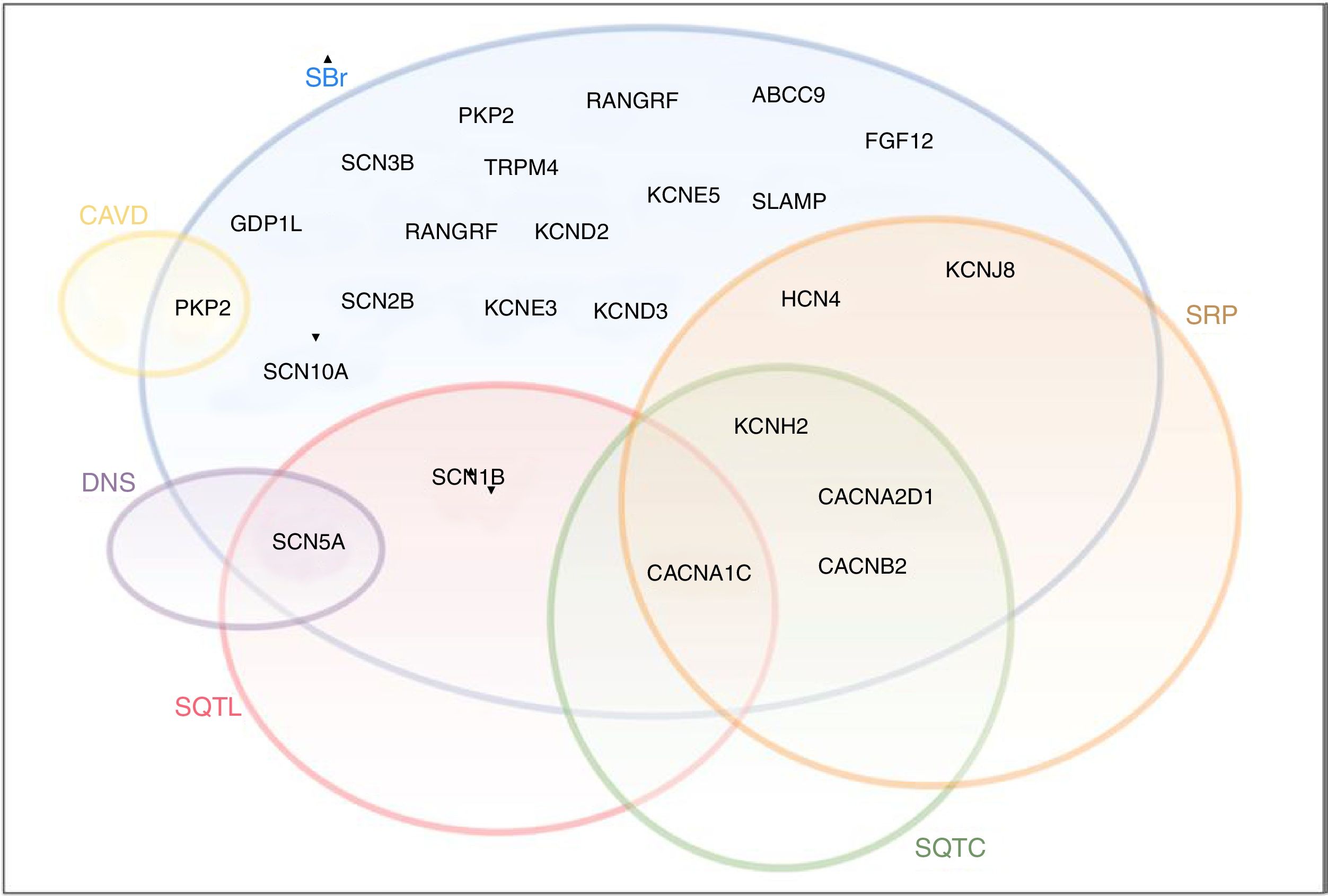
Diagrama ilustrativo do overlap entre SBr, SQTL, SQTC, DNS, SRP e CAVD. A negrito encontram‐se os genes do complexo macromolecular do canal de sódio.
CAVD: cardiomiopatia arritmogénica do ventrículo direito; DNS: doença do nó sinusal; SQTC: síndrome do QT curto; SQTL: síndrome do QT longo; SRP: síndrome de repolarização precoce.
Retirado e adaptado de Sarquella‐Brugada (2016)8 e Fernandez‐Falgueras (2017).35
Além disso, recentemente, alguns estudos reportam anomalias cardíacas estruturais secundárias a mutações nesse gene (nomeadamente CMD), embora se desconheça o mecanismo subjacente.6,18,22,77.
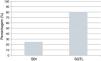
Atualmente, o poder dos testes genéticos para identificar mutações é de 25% na SBr e 80% na SQTL (Figura 9)43,78. O impacto da genética na abordagem clínica varia consideravelmente de acordo com a canalopatia subjacente e é mais marcado na SQTL, na qual se reconhece influência ao nível do diagnóstico, prognóstico e terapêutica9,43,78.
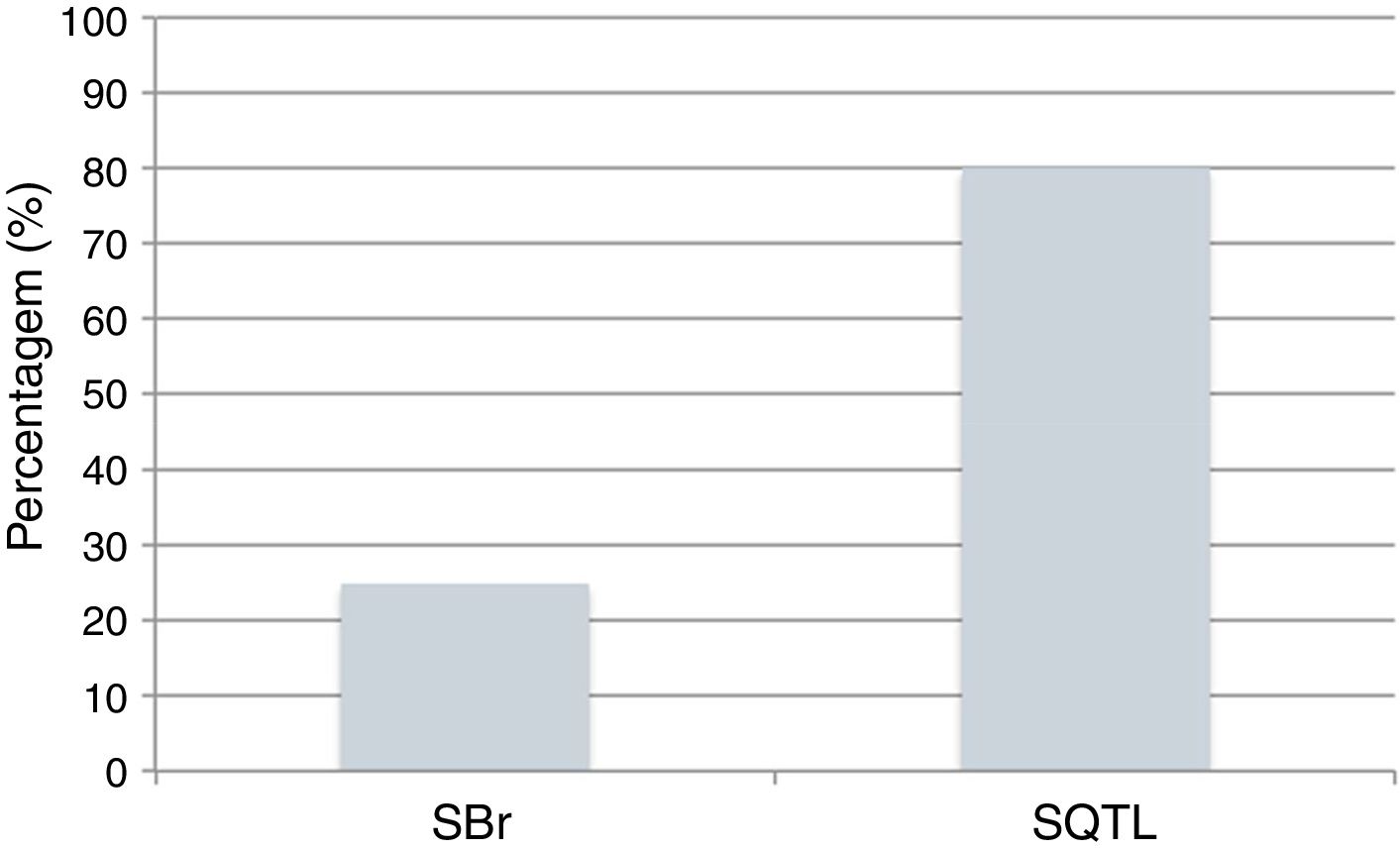
Canalopatias cardíacas: positividade dos testes genéticos em indivíduos clinicamente diagnosticados com SBr e SQTL.
Dados retirados de Schwartz e Dagradi (2016).78
Grandes progressos têm sido feitos na compreensão da relação genótipo‐fenótipo e suas implicações43. Todavia, apesar da evolução do conhecimento científico nessa área, o genótipo de um número considerável de indivíduos afetados permanece indeterminado, alguns mecanismos continuam por esclarecer e as opções terapêuticas disponíveis são, ainda, limitadas12,18,72. A melhor compreensão dos fundamentos moleculares poderá contribuir não só para aprofundar esses últimos aspetos, mas também para o desenvolvimento de novas abordagens terapêuticas específicas para o gene ou a mutação12,43.
