



Syncope and palpitations as the only initial manifestations of myotonic dystrophy type 1 (MD1) due to a CTG expansion of 50–100 repeats have not been reported.
Case reportIn a 55-year-old female with a family history of MD1 and a personal history of a single syncope, palpitations, and hyperCKemia, 70 CTG repeats were detected in the DMPK gene. Her brother had presented atypical clinical, electromyographic, and muscle biopsy features since the age of 35 but had been diagnosed with MD1 after he later developed typical distal myotonia. He died suddenly during an episode of syncope at the age of 53. A sister with clinical myotonia died suddenly during sleep at the age of 45 and a second sister with quadriparesis died from complications of intestinal rupture at age 52. A third sister committed suicide at age 40 after developing recurrent syncopes, while a fourth sister had hyperCKemia and foot-extensor weakness. The mother of these five affected children died suddenly from myocardial rupture.
ConclusionsMD1 with <100 CTG repeats may exclusively manifest cardiologically. Family screening for MD1 is important even in asymptomatic patients. MD1 may initially manifest without typical features, while muscle biopsy may be misleading and indicate glycogenosis. Close cardiac follow-up is important if MD1 manifests cardiologically to prevent syncope or sudden cardiac death.
Na distrofia miotónica tipo 1 (DM1) devido a expansão CTG 50-100 não foram reportadas até ao momento síncope e palpitações como manifestações iniciais da mesma.
Caso clínicoNuma mulher de 55 anos com história familiar de DM 1 e antecedentes de um único episódio síncopal, palpitações e hiper CKemia, foi detetada uma expansão de CTG-repeat de 70 no gene DMPK. O irmão apresentava desde os 35 anos características clínicas, eletromiográficas e nas biópsias musculares atípicas tendo-lhe sido diagnosticada DM 1 após ter desenvolvido mais tarde miotonia distal típica. Morreu subitamente no contexto duma síncope aos 53 anos. Uma irmã com miotonia clínica morreu subitamente aos 45 anos durante o sono. Uma segunda irmã com quadriparesia morreu de complicações de rotura do intestino aos 52 anos. Uma terceira irmã cometeu suicídio aos 40 anos após ter desenvolvido síncopes recorrentes. Uma quarta irmã tinha hiper CKemia e fraqueza muscular nos pés. A mãe destes 5 filhos afetados morreu subitamente de rotura do miocárdio.
ConclusãoA DM1 com CTG-repeat expansão < 100 pode manifestar-se exclusivamente do ponto de vista cardiológico. O rastreio familiar para DM1 é importante mesmo nos doentes assintomáticos. A DM1 pode manifestar-se inicialmente sem características típicas de DM1. A biópsia muscular na DM1 pode ser enganadora e indicar glicogenose. Um seguimento cardíaco rigoroso é importante se a DM1 se manifesta sob o ponto de vista cardiológico para prevenir a síncope ou morte súbita.
Clinical manifestations in patients with myotonic dystrophy type 1 (MD1) carrying a CTG expansion of 50–100 repeats are usually mild and include ptosis or cataract.1–3 Syncope has occasionally been described as a manifestation of MD14,5 but syncope and palpitations as the only initial manifestations of MD1 due to a CTG expansion of 50–100 repeats have not been reported.
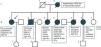
Case reportThe index patient (II/1) is a 55-year-old female, height 165 cm, weight 58 kg, with a history of a single syncope four years earlier and recurrent early morning palpitations since then, treated by beta-blockers (Figure 1). She had a family history of MD1 and was referred for assessment of genetic status. She reported daytime sleepiness but her clinical neurologic and cardiological exam was completely normal. Blood tests, however, revealed hyperCKemia of 264 U/l (normal 26–145 U/l). Work-up for the syncope in 2014, including cerebral magnetic resonance imaging, carotid ultrasound, and electroencephalogram, was normal. Cardiologic examination including telemetry and echocardiography was uninformative. On standard ECG incomplete right bundle branch block was recorded once. Genetic testing by PCR revealed a heterozygous CTG expansion of 70 repeats in the DMPK gene.

Her family history was noteworthy for at least five sibs affected out of a total of six (Figure 1). Her brother (II/2) manifested at onset in 1991 (age 35) with reduced motility of the tongue and difficulty with chewing and closing his mouth. Following these abnormalities he developed myotonia of both hands, daytime sleepiness, easy fatigability, and adynamia. Starting in 1998 (age 42) he reported permanent tiredness and aching muscles after exercise. A clinical neurologic exam at that time revealed wasting of the tongue edges, tongue fasciculations, clinical myotonia, and wasting of the thighs. Blood tests revealed hyperlipidemia, mild hyperCKemia of 204 U/l (normal <172 U/l), and mild elevation of gamma-glutamyl-transpeptidase (57 U/l; normal <55 U/l). Ischemic exercise testing and lactate stress testing were normal. Needle electromyography (EMG) showed marked myopathic alterations but no spontaneous activity. Muscle biopsy revealed a myopathic syndrome with accumulation of fat and particularly glycogen, which was interpreted as indicative of glycogenosis (Figure 1). Biochemical investigations revealed normal activity of respiratory chain complexes. Visually evoked potentials were noninformative. Echocardiography was indicative of hypertrophic cardiomyopathy, the left ventricular myocardium having abnormal texture. The 24-hour ECG was normal. In 2004 (at age 48) he presented with mild dysarthria and clinical myotonia but no muscle weakness. This time needle EMG revealed myotonic discharges in the thenar muscles. MD1 was diagnosed genetically on detection of a heterozygous CTG repeat expansion of 500. Colonoscopy in 2006 (age 49 years) revealed a cecal tubular adenoma and gastroscopy in 2008 (at age 52) revealed Barrett esophagus. He died suddenly during an episode of syncope in 2009 at age 52.
Three sisters (II/3, II/4, and II/5) were also affected. One sister (II/3) manifested marked myotonia and died suddenly during sleep at age 45. A second (II/4) developed quadriparesis requiring a wheelchair, and experienced a spontaneous rupture of the intestines, dying from septic complications at age 52. In both these sisters (II/3 and II/4) the diagnosis was genetically confirmed but detailed results are no longer available. A third sister (II/5) experienced recurrent syncopes of unknown cause but without other symptoms or signs and committed suicide for unknown reasons at age 40. The 64-year-old fourth sister (II/6) had a history of recurrent hyperCKemia, hypothyroidism, endometriosis, hypertension, and right-sided foot extensor weakness since at least the age of 55 (Figure 2). The mother of these seven children (I/2) required treatment for high blood pressure. She experienced myocardial rupture leading to sudden cardiac death (SCD). The father (I/1) of the seven was symptom-free but died from lymphoma.

Neuropathology of the muscle biopsy. H&E staining (A) shows discrete fiber size variation, fiber splitting and internalized nuclei; Oil-red-O staining (B) shows increased lipid accumulation; PAS-staining (C) shows storage of excess glycogen; Excess glycogen deposits are also detectable at an ultrastructural level within and between the fibers (A-C) ×20 magnification (D) × 3000 magnification.
The presented family with MD1 is interesting for several reasons. Firstly, the index patient had a CTG repeat expansion <100 and manifested exclusively with cardiogenic syncope and hyperCKemia. Patients with 50–100 CTG repeats are frequently asymptomatic or develop only mild manifestations such as cataract, ptosis, or clinical myotonia.1,2 If these patients become symptomatic, onset is later in life, at ages ranging between 20 and 70 years.2 In a systematic study of 102 MD1 patients with small CTG expansions (50–99 CTG repeats), most patients were asymptomatic.3 Those who were symptomatic had developed cataracts, myotonia, excessive daytime sleepiness, or myotonic discharges on needle EMG.3 None of these patients had developed cardiac symptoms, but ECG abnormalities were recorded in 21%.3 In an 11-year-old girl with a CTG expansion of 91 repeats, MD1 manifested as myopathy,6 and a Japanese male with 60–70 CTG repeats developed mild weakness of the sternocleidomastoid muscle at age 66.7 In a study of 14 MD1 patients two had <100 CTG repeats, both of whom manifested with cataract and mild myotonia. Both had reduced myocardial Doppler velocities but no cardiac symptoms.8 Only in patients with small expansions is there a negative correlation between expansion size and age at onset.9
Secondly, presumably cardiac manifestations of MD1 resulted in the sudden death of two family members (II/2 and II/3). In patient II/2 no syncope had occurred until the one during which he died. Patient II/5 was living in Switzerland, which is why no information about the cause of her recurrent syncopes was available, but she did not have a central nervous system disorder. Since cerebral causes of syncope were largely excluded in all three sibs with syncope or SCD, the syncopes were attributed to a cardiac rather than a neurologic cause. Most likely, syncopes in patients II/1, II/2, and II/5 are attributable to cardiac arrhythmias, although confirmation by standard or long-term ECG is lacking. However, even small CTG expansions of 60–70 repeats may manifest with ECG abnormalities, such as an increased His-ventricular interval.7,8
Thirdly, the brother in whom MD1 was first diagnosed in this family (II/2) did not present with a myopathic face, frontal baldness, or limb weakness at onset and did not show spontaneous activity at any of the sites investigated on needle EMG. His difficulties in closing his mouth properly in 1998 may have been the beginning of facial manifestations, but he developed typical clinical features of MD1 during the following six years. Late onset of clinical manifestations (at the age of 35) and absence of a typical myopathic face may also be due to the relatively small CTG expansion in this patient. Absence of myotonic or pseudomyotonic discharges on EMG is not unusual and may depend on the recording site and disease duration.
Fourthly, muscle biopsy in the brother (II/2) of the index patient (II/1) revealed a marked increase in glycogen deposition, which is an unusual finding. Though increased lipid deposition has been occasionally reported in MD1,2 increased glycogen deposition is rare.10 Granula containing glycogen seem to be particularly increased in patients with congenital MD1.11 Interpretation of the muscle biopsy as indicative of glycogenosis was misleading and delayed the correct diagnosis.
This case shows that MD1 with <100 CTG repeats may exclusively manifest cardiologically, that family screening for MD1 is important even in asymptomatic patients, and that MD1 may initially manifest with atypical clinical features. Muscle biopsy in MD1 may be misleading and may indicate glycogenosis. Close cardiac follow-up is important if MD1 manifests cardiologically to prevent syncope or SCD.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflicts of interestThe authors have no conflicts of interest to declare.
