





A distrofia miotónica é uma doença multissistémica, que se associa a alterações cardíacas que são responsáveis por elevada taxa de morbilidade e mortalidade. O atingimento do tecido de condução é frequente, traduzindo‐se em alterações no ritmo cardíaco, que tendem a progredir com a idade.
ObjetivoO presente trabalho propõe‐se avaliar o perfil cardiovascular global, bem como o risco de arritmias cardíacas, em indivíduos com distrofia miotónica de Steinert tipo 1 (DMS1), correlacionando os diversos achados com o estudo genético (tamanho da expansão CTG) comparando possíveis diferenças entre géneros.
MétodosEstudo retrospetivo que incluiu 31 pacientes DMS1 referenciados ao serviço de cardiologia do Centro Hospitalar Entre Douro e Vouga pelo serviço de neurologia para rastreio de doença cardíaca. Todos os doentes realizaram uma consulta clínica e foram submetidos a exames complementares de diagnóstico, nomeadamente eletrocardiograma (ECG), eletrocardiograma de alta resolução (ECGAR), variabilidade da frequência cardíaca (VFC), monitorização eletrocardiográfica ambulatória contínua (MEAC‐Holter) e ecocardiograma transtorácico (ETT). Foi ainda consultado o exame genético previamente efetuado.
ResultadosDos 31 doentes estudados, 38% apresentaram BAV 1.° grau e 51% apresentaram perturbação da condução intraventricular no ECG; 62% dos doentes apresentaram potenciais ventriculares tardios; não se verificou doença cardíaca no ETT; as extrassístoles supraventriculares e ventriculares raras foram as arritmias mais frequentes no Holter de 24h; a amostra apresentou valores diminuídos para a VFC, o que reflete a presença de disfunção vagal. Os doentes com maiores expansões CTG apresentaram maior número de anormalidades cardíacas.
ConclusõesPacientes com DMS1 apresentam alterações cardíacas, embora a gravidade das alterações documentadas esteja aquém do sugerido na literatura. Este aspeto poderá refletir a qualidade do seguimento clínico oferecido aos doentes por um centro com uma consulta especializada.
Myotonic dystrophy is a multisystem disease associated with cardiac abnormalities that are responsible for high morbidity and mortality. It commonly affects conduction tissue, resulting in changes in heart rate that tend to progress with age.
ObjectiveThe aim of the study was to assess overall cardiovascular risk and the risk of arrhythmias in patients with myotonic dystrophy type 1 (DM‐1) and to correlate them with genetic study (CTG expansion size).
MethodsThis retrospective study included 31 DM‐1 patients referred to the cardiology department of Centro Hospitalar Entre Douro e Vouga by the neurology department for screening for heart disease. Patients’ medical records were consulted for the diagnostic tests performed in the diagnostic cardiology consultation: electrocardiogram (ECG), high‐resolution ECG, heart rate variability (HRV), Holter 24‐hour ambulatory ECG and transthoracic echocardiogram (TTE); results of genetic testing were also consulted.
ResultsOf 31 patients studied, 38% had first‐degree atrioventricular block (AVB) and 51% had intraventricular conduction disturbances (62% had late potentials). TTE revealed no structural heart disease. Rare supraventricular and ventricular ectopic beats were the most common arrhythmias on 24‐hour Holter monitoring. The sample showed lower HRV, reflecting vagal dysfunction. Patients with larger CTG expansions had more cardiac abnormalities.
ConclusionsPatients with DM‐1 had arrhythmic events, with AVB and more significantly intraventricular block, although none had malignant arrhythmias or structural heart disease. No patient died. Patients with larger CTG expansions had greater involvement of cardiac conduction tissue.
A distrofia miotónica tipo 1 (DMS1), ou doença de Steinert, é uma doença genética que tem como causa a expansão anormal do tripleto citosina, timina e guanina (CTG) no gene dystrophia myotonica‐protein kinase (DMPK), situado no cromossoma 19, tendo uma expressão importante no músculo‐esquelético e cardíaco1. A DMS1 é considerada a forma mais frequente de distrofia muscular nos adultos, com uma prevalência de 2‐14:100 000 indivíduos e uma incidência de 1:8.0002–5. É uma doença multissistémica, hereditária, de transmissão autossómica dominante, com penetrância incompleta.
Apesar de a DMS1 ter como principal manifestação clínica a miotonia e a fraqueza muscular, secundárias ao envolvimento que esta condiciona no músculo‐esquelético, outros órgãos e sistemas poderão ser afetados, nomeadamente o sistema cardiovascular e o sistema respiratório6. De facto, a reduzida esperança média de vida nestes doentes está fortemente dependente de elevadas taxas de mortalidade devido a infeções respiratórias, neoplasias e a morte súbita cardíaca (MS)5.
A doença cardíaca é assim comum na DMS1, embora a sua prevalência seja difícil de estimar com precisão. Dados indicam que as perturbações da condução são a manifestação cardíaca mais comum da DMS1, embora outras complicações tenham sido descritas, tais como taquiarritmias6,7. Por outro lado, existem dados que indicam que o agravamento do comprometimento muscular afeta a progressão dos eventos arrítmicos, podendo a prevalência destes eventos chegar aos 80% dos casos8. Para além disso, as arritmias cardíacas constituem a causa mais frequente de mortalidade na DMS1 depois da pneumonia, sendo responsável pela morte de até 30% dos pacientes. A grande maioria dos pacientes (mais de 60%) que morrem por patologia cardíaca tem morte súbita9.
Atendendo ao impacto que as manifestações cardíacas na DMS1 têm na sobrevida dos doentes, torna‐se importante identificar potenciais preditores de morte súbita, que permitam fazer uma adequada estratificação do risco cardiovascular destes doentes. Neste sentido, alguns autores defendem que, quer o grau de atingimento neurológico quer o tamanho da expansão CTG, estão diretamente relacionados com a gravidade da doença do tecido de condução cardíaco10–12. Por outro lado, a evidência de alterações severas no eletrocardiograma (ECG) tem sido indicado como um preditor independente para a morte súbita nestes doentes. No entanto, as características do envolvimento do tecido de condução na DMS1 e o facto de, inclusivamente, já terem sido constatadas alterações intermitentes da condução e ritmo num número significativo de doentes (32%) com ECG normal, coloca o ECG de 24h (Holter) como componente importante no seguimento destes doentes para a identificação e quantificação de arritmias5,13. As evidências relativas à potencial relevância do ECG de alta resolução (ECGAR) neste contexto demonstram‐se inconclusivas, com indicação de boa sensibilidade, mas baixa especificidade, para a presença de arritmias ventriculares3. Também as medidas de avaliação da modulação autonómica cardíaca (nomeadamente a variabilidade da frequência cardíaca – VFC) têm sido estudadas, não existindo, contudo, dados estabelecidos quanto ao seu valor preditivo para MS em doentes com DMS114. O ecocardiograma transtorácico (ETT) tem também sido usado para despiste de doença cardíaca nestes doentes, apesar de a cardiomiopatia ser muito rara nestes pacientes e os estudos disponíveis limitados na informação que têm veiculado a este respeito3.
O objetivo deste trabalho é avaliar o risco de disritmias em indivíduos com DMS1 comparando possíveis diferenças entre géneros e caracterizar o impacto da doença a nível cardíaco, correlacionando estas alterações com o estudo genético (tamanho da expansão CTG).
MétodosEstudámos retrospetivamente (entre 2000‐2011) 34 doentes com o diagnóstico de DMS1, referenciados pelo Serviço de Neurologia à Consulta de Cardiologia do Centro Hospitalar Entre Douro e Vouga, para rastreio cardíaco, no qual realizaram exames de diagnóstico (ECG, ECGAR, ETT, Holter 24h e VFC). A população em estudo incluiu 31 dos 34 doentes inicialmente englobados, devido à aplicação dos seguintes critérios de exclusão: outros tipos de miotonia ou DM e doentes que não completaram os protocolos da consulta. A idade variou de 18‐66 anos, sendo 11 doentes do sexo masculino e 20 doentes do sexo feminino.
Os doentes realizaram ECG de 12 derivações em condições standard15, tendo‐se extraído a seguinte informação: ritmo cardíaco; frequência cardíaca; eixo do QRS; intervalos PR, QRS, QT, QTc. Todos os ECG foram avaliados independentemente por dois observadores experientes, seguindo as recomendações internacionais15.
O ECGAR foi realizado também em condições standard16, tendo‐se extraído as seguintes variáveis: duração do complexo QRS filtrado (QRSd); a raiz quadrada média do sinal nos últimos 40ms do QRS (RMS40); duração dos sinais menores que 40mV no final do QRS (LAS). A existência de potenciais ventriculares tardios (PVT) foi assumida quando dois dos índices avaliados se encontrassem anormais, face aos critérios de normalidade assumidos: QRSd<114ms; RMS40>20μV e LAS<38ms)17.
O ETT foi também realizado em condições standard18, tendo‐se considerado para análise as seguintes variáveis: morfologia e função valvular; dimensões das cavidades cardíacas; espessura das paredes ventriculares; avaliação da função sistólica global ventricular esquerda: fração de ejeção (FE%); velocidade de encurtamento da fibra circunferencial (Vcf) (permite avaliar a contratilidade do miocárdio); índice de massa ventricular esquerda (IMVE).
Na análise e na interpretação do registo do Holter, realizado também em condições standard18 consideramos para análise as perturbações da condução/ritmo encontradas, adotando‐se a classificação de Lown para a caracterização da extrassistolia. Na análise da VFC, feita nos registos de Holter, os índices avaliados no domínio do tempo foram: desvio padrão dos intervalos R‐R (SDNN); desvio padrão dos intervalos R‐R calculados em intervalos de cinco minutos (SDANN); raiz quadrada da diferença ao quadrado média de intervalos R‐R sucessivos (RMSSD); proporção de números de pares de intervalos R‐R que diferem mais de 50ms, dividida pelo número total de intervalos R‐R (pNN50). No domínio da frequência foram avaliados: potência espetral nas altas frequências (HF); potência espetral nas baixas frequências (LF); razão LF/HF.
Para a análise estatística da correlação da expansão do CTG com os eventos cardíacos dividimos a amostra em dois grupos, em função da mediana das duplicações na amostra (mediana=900), com um grupo com valor de expansão inferior à mediana e o outro grupo com valor superior à mediana.
A análise estatística dos dados foi realizada utilizando o Software SPSS® versão 19.0. Procedeu‐se a uma análise descritiva das diferentes variáveis e à comparação de possíveis diferenças entre géneros. As variáveis quantitativas foram apresentadas como média±desvio padrão, e respetiva amplitude de variação, e as qualitativas em frequência absoluta.
Para a comparação da média das variáveis quantitativas analisadas para ambos os géneros da amostra e em função da expansão do CTG (cut‐off de 900 duplicações), foi utilizado o teste paramétrico t‐Student para amostras independentes. Para a análise de variáveis categóricas recorreu‐se aos testes qui‐quadrado e teste exato de Fisher, conforme apropriado.
Adotou‐se um valor de p<0,05 para um intervalo de confiança de 95% como critério de significância estatística.
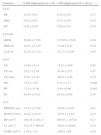
ResultadosCaracterização da amostraA amostra final foi constituída por 31 doentes, sendo 20 do sexo feminino (64,5%) e 11 do sexo masculino (35,5%), com uma idade média de 42,00±13,10 anos, variando entre os 18 e os 66 anos. O índice de massa corporal (IMC) médio foi de 27,14±4,72Kg/m2. Em termos comparativos, o grupo das mulheres apresentou um valor médio de IMC superior aos homens (29,36±5,11kg/m2versus 25,75±4,08kg/m2, respetivamente; p=0,04, conforme Tabela 1), diferença que se verificou também no que concerne ao peso, altura e superfície corporal (conforme Tabela 1).
Caraterização da amostra de doentes com DMS1 por grau de associação entre as variáveis em estudo e o género
| Variáveis | Total (n) | Mulheres (n=20) | Homens (n=11) | p |
| Idade (anos) | 31 | 43,35±13,42 | 39,55±12,7 | 0,44 |
| Peso | 31 | 70,60±11,94 | 71,00±10,32 | 0,92 |
| Altura | 31 | 155,20±3,69 | 166,27±4,94 | <0,001 |
| Sup. corporal | 31 | 1,69±0,12 | 1,78±0,11 | 0,05 |
| IMC | 31 | 29,36±5,11 | 25,75±4,08 | 0,04 |
No ECG, 19 doentes (61%) apresentaram ECG normal. Dos restantes, 12 doentes (38%) apresentavam BAV do 1.° grau, dos quais nove eram mulheres (29%) e três eram homens (9,7%). Em 16 doentes (51%) verificaram‐se perturbações da condução intraventricular, dos quais nove eram mulheres (29%) e sete eram homens (23%). A duração média do QRS foi de 0,11±0,20s, observando‐se uma duração significativamente maior deste nos doentes do sexo masculino (duração do QRS=0,12±0,20s) versus as doentes do sexo feminino (duração do QRS=0,1±0,21s; p=0,05, conforme Tabela 2).
Caráter
| Variáveis | Feminino (n=20) | Masculino (n=11) | p |
| ECG | |||
| PR (seg.) | 0,19±0,01 | 0,19±0,01 | 0,74 |
| QRS (seg.) | 0,10±0,21 | 0,12±0,20 | 0,05 |
| QT (seg.) | 0,40±0,02 | 0,40±0,04 | 0,66 |
| QTC (seg.) | 0,40±0,08 | 0,40±0,12 | 0,63 |
| Eixo QRS (°) | 44,45±44,78 | 35,63±45,21 | 0,60 |
| ECGAR | |||
| QRSd (ms) | 101,76±20,80 | 114,89±19,52 | 0,13 |
| RMS 40 (μV) | 14,79±10,86 | 16,02±11,64 | 0,79 |
| LAS 40 (ms) | 51,76±19,09 | 44,00±17,16 | 0,31 |
| ECO | |||
| AE (mm) | 35,10±4,97 | 35,18±3,43 | 0,96 |
| VE diást. (mm) | 45,40±3,83 | 47,55±4,90 | 0,18 |
| VE síst. (mm) | 29,55±5,02 | 30,55±4,15 | 0,58 |
| SIV (mm) | 8,50±2,01 | 9,45±1,63 | 0,18 |
| PP (mm) | 7,65±1,13 | 8,36±1,02 | 0,09 |
| FE (%) | 64,33±11,03 | 65,82±5,64 | 0,68 |
| Vcf (circumf./s) | 35,10±7,42 | 35,93±4,02 | 0,73 |
| IMVE (g/m2) | 59,48±13,00 | 66,23±10,74 | 0,15 |
| RWT (mm) | 0,33±0,05 | 0,35±0,51 | 0,39 |
| VFC | |||
| SDNN (ms) | 23,27±14,27 | 17,88±14,97 | 0,18 |
| SDANN (ms) | 18,16±11,26 | 16,88±10,86 | 0,31 |
| RMSSD (ms) | 38,37±22,47 | 35,09±25,64 | 0,71 |
| PNN50 (%) | 17,47±14,97 | 13,18±17,58 | 0,48 |
| HF (mV2) | 732,81±560,00 | 296,8±110,50 | 0,32 |
| LF (mV2) | 422,55±460,88 | 438,6±420,66 | 0,94 |
| LF/HF (mV2) | 1,07±1,04 | 2,95±1,64 | 0,007 |
| Holter | |||
| ESV | 14 (45%) | 8 (26%) | 1 |
| EV | 11 (36%) | 6 (19,4) | 0,7 |
| FA | 1 (3%) | 0 | 1 |
| Bloqueios | 8 (26%) | 5 (16%) | 0,4 |
O valor do intervalo QT e QTC apresentaram‐se normais na nossa amostra, não se verificando diferenças estatisticamente significativas entre os géneros (p>0,05).
A maior parte da amostra (87%) apresentou eixo elétrico normal, com apenas três doentes do sexo feminino (15%) e um do sexo masculino (9%) a apresentarem desvio direito do eixo elétrico.
Eletrocardiograma de alta resoluçãoNuma análise geral do ECGAR, dez doentes (38%) apresentaram um ECGAR normal, enquanto 16 doentes (62%) apresentaram PT positivos, sendo de salientar que os índices que se apresentaram aumentados na maior parte da amostra foram o RMS 40 e o LAS 40.
Ecocardiograma transtorácicoRelativamente à avaliação ecocardiográfica, não se encontraram alterações significativas nos diversos índices ecocardiográficos estudados. Em termos estruturais, encontraram‐se dois doentes do sexo feminino (7%) com dilatação ligeira da AE e seis doentes (20%) apresentaram hipertrofia ligeira do SIV, dos quais três do sexo feminino (10%) e três do sexo masculino (10%). A dimensão do ventrículo esquerdo (VE) em sístole e em diástole foi normal em toda a amostra, bem como o IMVE e a Vcf. A fração de ejeção ventricular esquerda (FEVE) estava ligeiramente diminuída em três doentes do sexo feminino (10%).
Na classificação da geometria do VE verificou‐se que a maior parte dos doentes (97%) apresentavam uma geometria do VE normal. Apenas um doente (3%) do sexo feminino apresentou remodelação concêntrica, pois a massa do VE estava normal, existindo porém um aumento da espessura relativa das paredes. Nenhum doente apresentou alterações morfofuncionais valorizáveis.
HolterRelativamente à avaliação no Holter, verificou‐se que o grupo das mulheres apresentava uma carga arrítmica ligeiramente maior (ESV=45% e EV=36%) do que a do grupo dos homens (ESV=26% e EV=19%). Contudo, não foram documentadas formas complexas de taquidisritmia em nenhum dos doentes estudados.
Apenas um doente apresentou um episódio de fibrilhação auricular paroxística.
Variabilidade da frequência cardíacaDa análise da VFC verificou‐se que a maioria da amostra (84%, n=26) apresentava diminuição da VFC, particularmente no sexo feminino (63 versus 37% nos homens). Os índices SDNN e SDANN encontraram‐se diminuídos em 18 mulheres (62% p=0,38) e em 11 homens (38% p=0,42). Por seu lado, considerando os parâmetros da análise no domínio da frequência, os homens apresentaram valores mais baixos de HF em relação às mulheres, bem como um aumento da relação LF/HF, revelando, em termos relativos, uma maior ativação simpática (conforme Tabela 2).
Tamanho da expansão de citosina, timina e guanina e perfil cardíacoPara avaliar o perfil cardiovascular em função da extensão da expansão do CTG estimou‐se a mediana desta e dividiu‐se a amostra em dois grupos: um com valor de expansão da CTG inferior a 900 (mediana) e outro com valor superior ou igual a 900. Em termos de características gerais, não se encontraram diferenças estatisticamente significativas no que concerne à distribuição por género, idade e IMC (conforme Tabela 3).
Caraterização da amostra de doentes com DMS1 por grau de associação entre as variáveis em estudo e o género
| Variáveis | ≤900 duplicações (n=15) | >900 duplicações (n=16) | p |
| Sexo (n, %) | |||
| Masculino | 6, 40,00% | 5, 31,25% | |
| Feminino | 9, 60,00% | 11, 68,75% | 0,61 |
| Idade (anos) | 39,40±15,38 | 44,44±10,46 | 0,29 |
| Peso (kg) | 69,07±12,021 | 72,31±10,55 | 0,43 |
| Sup. corporal (m2) | 1,72±0,13 | 1,72±0,12 | 0,99 |
| IMC (kg/m2) | 26,61±5,36 | 29,45±4,40 | 0,11 |
No ECG de 12 derivações não se encontraram diferenças estatisticamente significativas (p>0,05) na amostra, embora quando comparado o subgrupo de doentes com número superior a 900 duplicações verificou‐se que estes apresentavam valores ligeiramente superiores nos intervalos PR e QT em relação ao grupo dos doentes com menos de 900 duplicações. A duração do QRS foi também significativamente maior no grupo de doentes com maior número de duplicações (0,12±0,02ms versus 0,10±0,2ms; p=0,02).
No ECGAR, o grupo dos doentes com maior número de duplicações de CTG apresentou valores médios superiores dos diversos parâmetros, atingindo significado estatístico para o QRSd e o LAS 40 (conforme Tabela 4).
Caraterização da amostra de doentes com DMS1 por grau de associação entre as variáveis em estudo e o género
| Variáveis | ≤900 duplicações (n=15) | >900 duplicações (n=16) | p |
| ECG | |||
| PR | 0,18±0,03 | 0,20±0,03 | 0,17 |
| QRS | 0,10±0,02 | 0,12±0,02 | 0,02 |
| QT | 0,39±0,26 | 0,40±0,03 | 0,30 |
| ECGAR | |||
| QRSd | 98,86±17,05 | 115,00±22,40 | 0,04 |
| RMS 40 | 18,41±11,39 | 11,49±9,47 | 0,10 |
| LAS 40 | 42,14±15,10 | 57,17±19,36 | 0,03 |
| ECO | |||
| AE | 34,40±4,15 | 35,81±4,69 | 0,38 |
| VE sist. | 29,27±5,16 | 30,50±4,27 | 0,47 |
| VE diast. | 45,67±4,30 | 46,63±4,36 | 0,54 |
| SIV | 7,87±1,59 | 9,75±1,77 | 0,004 |
| PP | 7,33±51,04 | 8,44±0,96 | 0,005 |
| FE | 65,84±10,81 | 63,93±8,09 | 0,58 |
| VFC | |||
| RMSSD (ms) | 37,43±22,88 | 36,94±24,40 | 0,61 |
| PNN50 (50%) | 16,64±16,79 | 15,25±15,44 | 0,38 |
| HF (mV2) | 450,78±418,17 | 659,91±155,10 | 0,11 |
| LF (mV2) | 422,55±460,88 | 438,6±420,66 | 0,10 |
| LF/HF (mV2) | 1,48±1,11 | 2,06±1,99 | 0,04 |
No ETT não foram observadas diferenças significativas entre os grupos quanto às dimensões da AE, SIV, VE em sístole e em diástole e ao valor da FE% (p>0,05).
No Holter, não se verificarem diferenças estatisticamente significativas entre os grupos.
Relativamente à VFC, verificou‐se que o grupo dos doentes com número superior a 900 duplicações (54%; p=0,88) apresentava VFC diminuída em relação ao grupo dos doentes com menos de 900 duplicações (31%; p=0,37).
DiscussãoAtendendo à baixa prevalência desta patologia, a série estudada representa um número elevado de doentes, assumindo‐se como uma amostra significativa e com características que permitem a exploração de dados, podendo os nossos achados ser um contributo na abordagem cardíaca destes doentes.
Numa primeira análise, verificamos que dois doentes da amostra implantaram pacemaker definitivo, como medida terapêutica, por bloqueio bifascicular sintomático. É também de realçar que nenhum doente desta amostra foi vítima de morte súbita.
Segundo a literatura, a DMS1 é mais comum na idade adulta, aspeto verificado também na nossa amostra, com a maioria dos doentes a apresentar uma idade entre os 30 e os 40 anos, sendo também doentes maioritariamente de estatura baixa e com excesso de peso, podendo este aspeto dever‐se às limitações motoras inerentes à mitonia e fraqueza muscular que caracterizam a doença.
Relativamente aos achados elétricos e ao contrário de outros registos19,20, na nossa série de casos encontrou‐se uma proporção relativamente baixa de doentes com BAV do 1.° grau no ECG (apenas 38%). Quanto à duração do complexo QRS, verificamos uma prevalência elevada de doentes com alargamento do QRS (cerca de 51% da série), em concordância com dados previamente publicados5, indicando um atingimento importante do tecido de condução intraventricular. Este aspeto pode também relacionar‐se com a evidência de uma elevada prevalência de doentes com PVT (cerca de 62%), em linha com resultados prévios indicando uma elevada prevalência de ECGAR anormais nestes doentes (cerca de 83% no estudo de Melacini et al.21), refletindo a existência de áreas de condução lenta e fragmentada no miocárdio destes doentes, constituindo um substrato potencial para o estabelecimento de arritmias ventriculares por reentrada. Contudo, e apesar de este exame ser sensível para a presença de arritmias ventriculares nestes doentes, tem uma especificidade extremamente baixa, comportando portanto um elevado número de falsos‐positivos3, aspeto aliás reforçado pela inexistência de formas complexas ou graves de arritmias ventriculares ou casos de morte súbita nos doentes em estudo.
Da observação global dos dados, um aspeto que também se destacou foi a constatação de um maior atingimento em termos de consequências físicas da doença, no género feminino, acompanhando um perfil cardiovascular igualmente mais desfavorável nas mulheres, aspeto que não tem sido sistematicamente apontado por outros estudos semelhantes.
Em termos estruturais, existe alguma controvérsia na literatura, com alguns autores a sustentarem o argumento de que existem alterações ecocardiográficas importantes nestes doentes, tais como o prolapso da válvula mitral, alterações da motilidade das paredes do VE, diminuição da FE%, diminuição da fração de encurtamento, hipertrofia e dilatação do VE, alterações na contratilidade segmentar e diminuição da função sistólica12,19,22. No nosso estudo não foram encontradas alterações ecocardiográficas significativas. De facto, a maior parte da amostra (94%) apresentou as diversas dimensões avaliadas dentro da normalidade, verificando uma hipertrofia ligeira do SIV em apenas 20% da amostra e apenas um doente apresentou FE% ligeiramente diminuída.
Outro aspeto que tem sito apontado como comum nestes doentes é a alteração do balanço autonómico, traduzida em grande medida por disfunção vagal5. De facto, os nossos dados indicam expressivamente uma proporção elevada de doentes com alterações nos diversos parâmetros da VFC, com particular relevância para os parâmetros espetrais (LF e HF e a sua relação). Curiosamente, as mulheres apresentaram valores superiores da banda HF e inferiores da banda LF e da relação LF/HF em relação aos homens, refletindo um maior predomínio relativo da atividade vagal em relação aos homens. Estes resultados sugerem diferenças dependentes do género, na modulação autonómica cardíaca nestes doentes, com maior atenuação da atividade vagal nos doentes do sexo masculino, o que poderá traduzir uma deterioração mais acelerada da função barorrecetora nestes doentes.
Um aspeto que tem sido também recorrentemente estudado na literatura prende‐se com a eventual dependência das complicações cardíacas em relação ao tamanho do tripleto CTG nos doentes com DMS15,8,19. As associações encontradas neste trabalho, e já relatadas em outros estudos, reforçam a existência de uma relação entre a gravidade genética e as alterações cardíacas. De facto, a presença de BAV do 1.° grau e BCRE no ECG, a presença de PT anormais, a presença de anormalidades no Holter e a diminuição da VFC esteve relacionada de forma significativa com o aumento do número de duplicações genéticas (tamanho do tripleto CTG). Desta forma, e com base nas evidências disponíveis, poderemos sugerir que o tamanho da expansão do CTG constitui uma importante forma de estratificação de risco de desenvolvimento de alterações cardíacas, embora a sua real importância careça de demonstração em estudos de seguimento alargado. Há que realçar, no entanto, que apesar dos nossos resultados não evidenciarem um risco arrítmico particularmente importante nesta série de doentes, é importante fazer um seguimento apertado nos doentes em risco de progressão, já que, de acordo com as guidelines, estes constituem indicação de classe IIb para a colocação de PCM/CDI23.
Considerando as limitações do presente trabalho, o fator mais determinante será seguramente o seu carácter retrospetivo. Atendendo a que a DMS1 é uma doença rara, a dimensão da amostra estudada é maior que o habitual, embora a potência dos testes estatísticos aplicados nestes contextos fique naturalmente comprometida.
ConclusãoNa realização deste trabalho incluíram‐se uma série de casos que representam parte dos doentes com DMS1 e que devido a um protocolo existente no Centro Hospitalar Entre Douro e Vouga foram observados na consulta de cardiologia para rastreio de doença cardíaca. Os achados deste estudo demonstram que existem alterações cardíacas em doentes com DMS1, embora a gravidade das alterações encontradas esteja aquém do sugerido na literatura. Este aspeto poderá refletir a qualidade do seguimento clínico oferecido aos doentes por um centro com uma consulta especializada para esta doença em concreto.
Responsabilidades éticasProteção de pessoas e animaisOs autores declaram que os procedimentos seguidos estavam de acordo com os regulamentos estabelecidos pelos responsáveis da Comissão de Investigação Clínica e Ética e de acordo com os da Associação Médica Mundial e da Declaração de Helsinki.
Confidencialidade dos dadosOs autores declaram ter seguido os protocolos do seu centro de trabalho acerca da publicação dos dados de pacientes.
Direito à privacidade e consentimento escritoOs autores declaram ter recebido consentimento escrito dos pacientes e/ou sujeitos mencionados no artigo. O autor para correspondência deve estar na posse deste documento.
Conflito de interessesOs autores declaram não haver conflito de interesses.
