







Failure of the normal cardiopulmonary transition can lead to persistent pulmonary hypertension of the newborn. Some degree of pulmonary hypertension complicates the course of more than 10% of all neonates with respiratory failure. This review article discusses the vascular abnormalities associated with neonatal pulmonary hypertension. Common therapies including inhaled nitric oxide, high frequency ventilation, surfactant, and extracorporeal life support are included. Promising new treatment approaches like phosphodiesterase inhibition and cell-targeted therapies are presented.
Uma falha na transição cardiopulmonar do feto para a vida extrauterina, pode levar ao desenvolvimento de hipertensão pulmonar persistente do recém-nascido. Mais de 10% de todos os recém-nascidos com insuficiência respiratória apresentam algum grau de hipertensão pulmonar. Neste artigo de revisão as alterações vasculares que estão associadas com a hipertensão pulmonar neonatal são apresentadas. As terapias mais comuns, como o óxido nítrico inalado, a ventilação de alta frequência, o surfactante e terapias de suporte de vida são revistas. Novas estratégias de tratamento, como a inibição de fosfodiesterases e a terapia celular direcionada são revistas.
Pulmonary hypertension (PH) presenting in the neonatal period may result from a number of causes, the most common of which is persistent pulmonary hypertension of the newborn (PPHN), caused by failure of normal cardiopulmonary transition.1,2 This syndrome is characterized by marked PH that causes hypoxemia and right-to-left extrapulmonary shunting of blood flow.3 PPHN is frequently associated with pulmonary parenchymal abnormalities including meconium aspiration, pneumonia and sepsis, and with pulmonary hypoplasia, maladaptation of the pulmonary vascular bed postnatally as a result of perinatal stress, or maladaptation of the pulmonary vascular bed in utero from unknown causes.4
This review focuses on recent progress in understanding of the pathophysiology and the treatment of neonatal PH.
Fetal pulmonary hypertension and the normal cardiopulmonary transitionFetal circulation is characterized by low systemic vascular resistance, elevated pulmonary vascular resistance (PVR)5–7 and the presence of shunts (Figure 1).
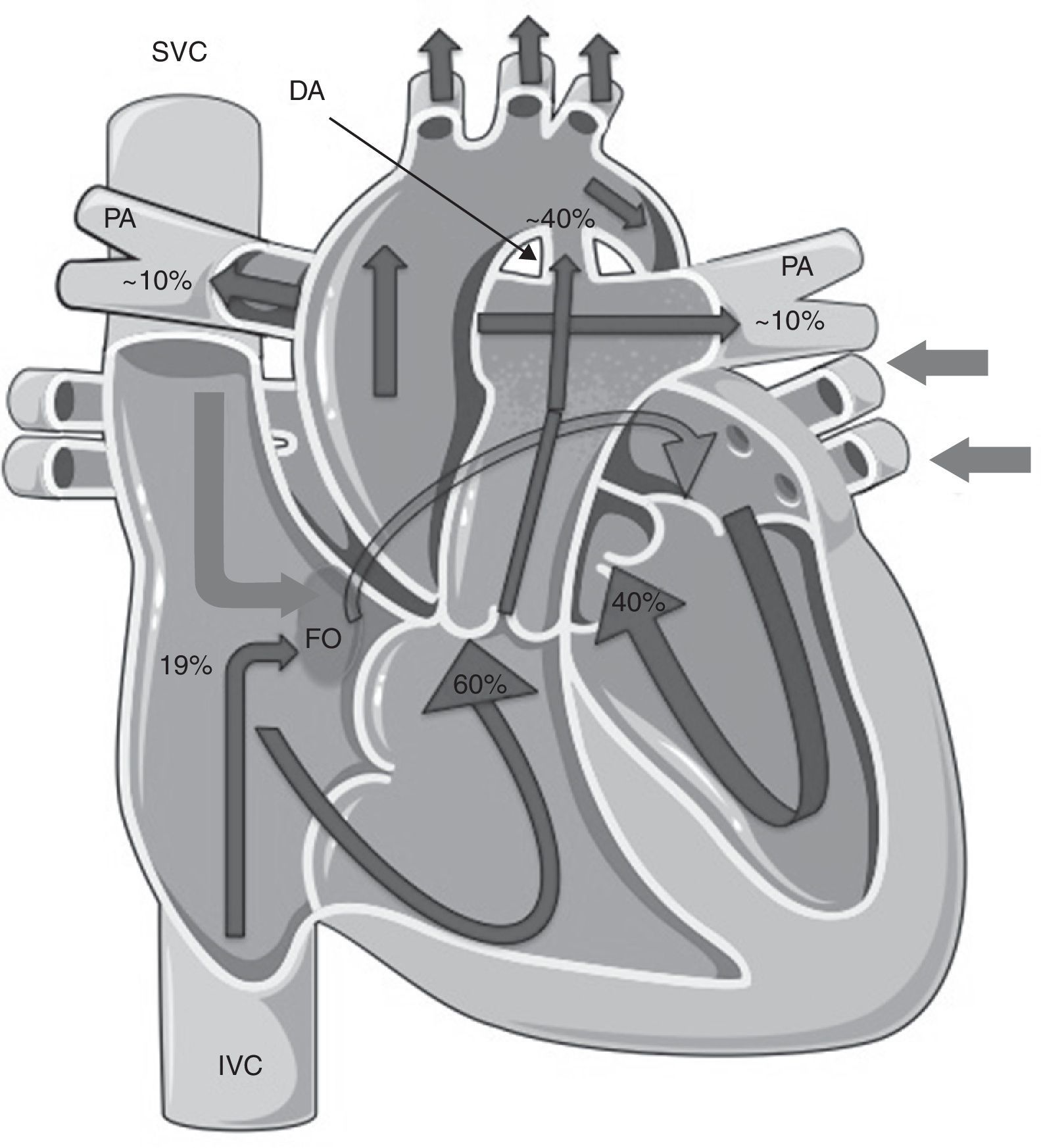
Fetal circulation. Note the presence of the ductus arteriosus and the foramen ovale and the distribution of blood flow, with the pulmonary arteries only receiving ∼10% of the total blood flow to the pulmonary circulation. Lighter arrows represent venous blood and darker (outlined) arrows represent arterial blood. DA: ductus arteriosus; FO: foramen ovale; PA: pulmonary arteries. (adapted) Copyright© 2012, Les Laboratoires Servier.
Numerous pathways appear to be involved in maintaining high pulmonary vascular tone preceding birth. Factors involved in elevated pulmonary resistance in the fetus include (i) the existence of a thick muscle layer in fetal pulmonary arterioles (mainly acquired during the last trimester of pregnancy),8 (ii) pulmonary vascular vasoconstriction induced by low O2 saturation, and (iii) dominance of vasoconstrictor vascular tone by substances like prostaglandins, leukotrienes, thromboxane and endothelin, and low production of vasodilators such as prostacyclin and nitric oxide (NO).9
The prostacyclin pathway has an important role in vascular tone. Cyclooxygenase is the rate-limiting enzyme that generates prostacyclin from arachidonic acid.10 The cyclooxygenase pathway also stimulates production of vasodilator prostaglandins and the vasoconstrictor thromboxane A2 (TXA2). Lipoxygenase metabolites of arachidonic acid, particularly leukotriene D4, may also contribute to elevated fetal PVR.3,11
Fetal pulmonary vessels have the ability to autoregulate flow through a myogenic response. This may explain why stimuli such as ductal compression, NO and increased oxygen tension cause only a transient increase in fetal pulmonary blood flow.11
Following delivery, the circulatory system must switch from fetal to neonatal circulation.7 When the umbilical cord is clamped, the low vascular resistance placental circuit is removed, resulting in a rise in neonatal systemic blood pressure. At the same time, lung expansion reduces both PVR and pulmonary artery pressure.7,8 These two changes decrease the right-to-left shunt at the ductus arteriosus, resulting in increased blood flow through the pulmonary arteries and lungs. With increased lung perfusion and expansion, neonatal oxygenation saturation is increased, and the ductus arteriosus begins to close.12 Ductus arteriosus closure is normally completed by 24 hours, but it may remain patent in infants with PH.9
As left atrial pressure increases and right atrial pressure falls, right-to-left shunting across the foramen ovale decreases.8
The production of prostacyclin (PGI2) is related to pulmonary shear stress3,9 and, although it is not essential for maintaining low PVR, PGI2 participates in the decline in PVR after the onset of ventilation at birth.
The fetus starts preparing for this shift late in gestation, increasing pulmonary expression of NO synthases and soluble guanylate cyclase. In the perinatal pulmonary vasculature, the effects of NO are primarily mediated by elevation of cyclic guanosine monophosphate (cGMP) via activation of soluble guanylyl cyclase and subsequently of cGMP-dependent protein kinase.3 The importance of the NO-cGMP pathway in normal transition has been demonstrated by acute or chronic inhibition of endothelial nitric oxide synthase (eNOS) in fetal lambs,13 which induces neonatal PH. Infants with PPHN have low plasma concentrations of arginine and NO metabolites,1 suggesting eNOS dysfunction.14
Decreased production of endogenous vasoconstrictors such as endothelin 1 (ET-1) and TXA2 is another mechanism that may contribute to the decline in PVR at birth. When subjected to various stimuli such as shear stress, hypoxia, or ischemia, ET-1 is transcripted, synthesized, and secreted within minutes. ET-1 binds primarily to the ETA receptor, and under physiologic conditions, ET-1 causes vasoconstriction by elevating intracellular calcium and sensitizing myofilaments to calcium.3 Circulating ET-1 levels in infants with PPHN are markedly elevated, correlate with disease severity and decline with resolution of PPHN.9 Because TXA2 is both a vasoconstrictor and a potent stimulus for platelet aggregation, it may be an important mediator of PH.15 In some infants with PPHN other vasoconstrictors, such as the leukotrienes LTC4 and LTB4, are also elevated.9
When the ductus arteriosus, ductus venosus and foramen ovale close during the transition to postnatal circulation, pulmonary vascular tone is dominated by vasodilator factors.10 Within minutes of this vasodilator response, increased pulmonary blood flow distends the vasculature, causing a structural reformation of the vascular wall, with flattening of the endothelium and thinning of smooth muscle and extracellular matrix.5
Pathophysiology of persistent pulmonary hypertension of the newbornWhen pulmonary vascular resistance does not fall after birth, hypoxemic respiratory failure or PPHN may result. PPHN is characterized by increased pulmonary vascular resistance and right-to-left shunting through the foramen ovale, with or without a patent ductus arteriosus, causing arterial hypoxia even with 100% FiO2.8
The main hallmarks of PPHN include sustained elevation of PVR, abnormal vasoreactivity, and structural remodeling of the pulmonary vascular bed.5 It is characterized as one of three types (Table 1): (i) underdevelopment of lung parenchyma (e.g., congenital diaphragmatic hernia); (ii) maladaptation of the pulmonary arteries due to lung parenchymal diseases such as meconium aspiration syndrome, respiratory distress syndrome, or pneumonia; and (iii) maladaptation of the pulmonary vascular bed, with normal lung parenchyma (such as perinatal asphyxia and idiopathic PPHN).1,16–19 Idiopathic PH is found in 10–20% of all infants with PPHN.1,20
Causes of persistent pulmonary hypertension of the newborn.
| Hypoplastic pulmonary vasculatureCongenital diaphragmatic herniaPulmonary hypoplasia |
| Abnormally constricted pulmonary vasculatureMeconium aspiration syndromePneumoniaRespiratory distress syndrome |
| Structurally abnormal pulmonary vasculatureIdiopathic persistent pulmonary hypertension |
Various risk factors have been proposed, including in utero exposure to nonsteroidal anti-inflammatory drugs and selective serotonin-reuptake inhibitors.21–23
Neonatal respiratory and heart diseases which cause hypoxemia or acidosis may precipitate pulmonary vasospasm, decreased pulmonary blood flow, increased right ventricular work and right-to-left shunting. The latter occurs through the ductus arteriosus, ductus venosus, and foramen ovale and intraparenchymally, maintaining high PVR and low lung compliance, creating a vicious circle in which reactive vasoconstriction intensifies the right-to-left shunt and the level of hypoxemia, thus preventing the postnatal reduction of PVR.1,24
Several neurohumoral factors are reported to be altered in PPHN (Figures 2 and 3). Decreases in eNOS gene and protein expression may limit perinatal NO production, thereby contributing to the pathophysiology of PPHN.3,24 Circulating endothelin levels are elevated in human infants with PPHN, while cGMP concentrations are reduced.24 Hypertension downregulates vascular endothelial growth factor (VEGF) expression in the developing lung, and this impaired VEGF signaling may contribute to the pathogenesis of PPHN.3 Phosphodiesterases (PDEs) have an important role in regulating cGMP and cyclic adenosine monophosphate (cAMP) in vascular smooth muscle. Consequently, PDE inhibitors like sildenafil and milrinone are the focus of research into new therapeutic approaches for PPHN that is resistant to traditional therapy with inhaled NO (iNO).10
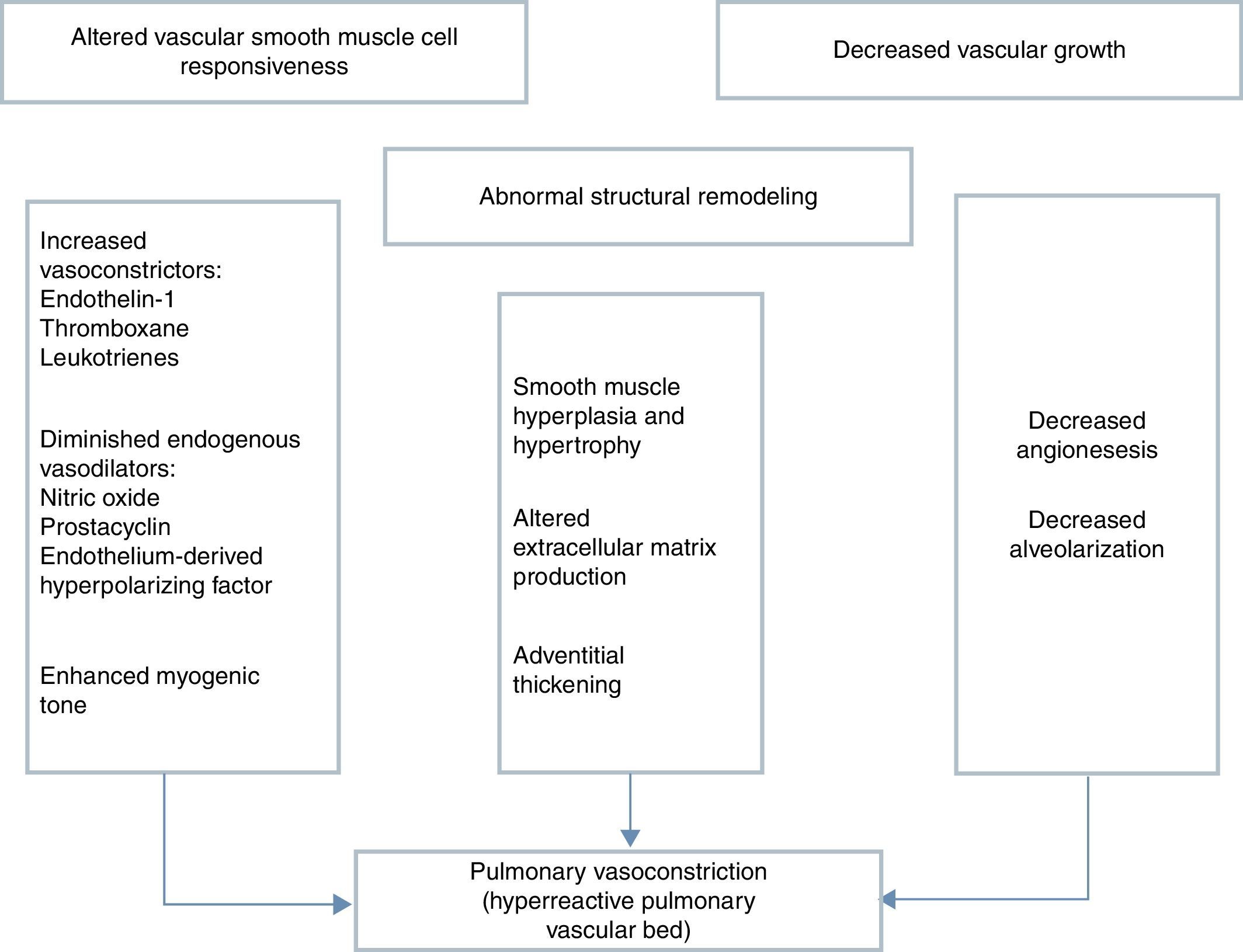
Mechanisms involved in the pathophysiology of persistent pulmonary hypertension of the newborn, which is characterized by abnormal vascular reactivity, cellular proliferation and vascular remodeling. The overall effect is compromised pulmonary vasodilation, severe central hypoxemia and extrapulmonary shunting of blood away from the lung.
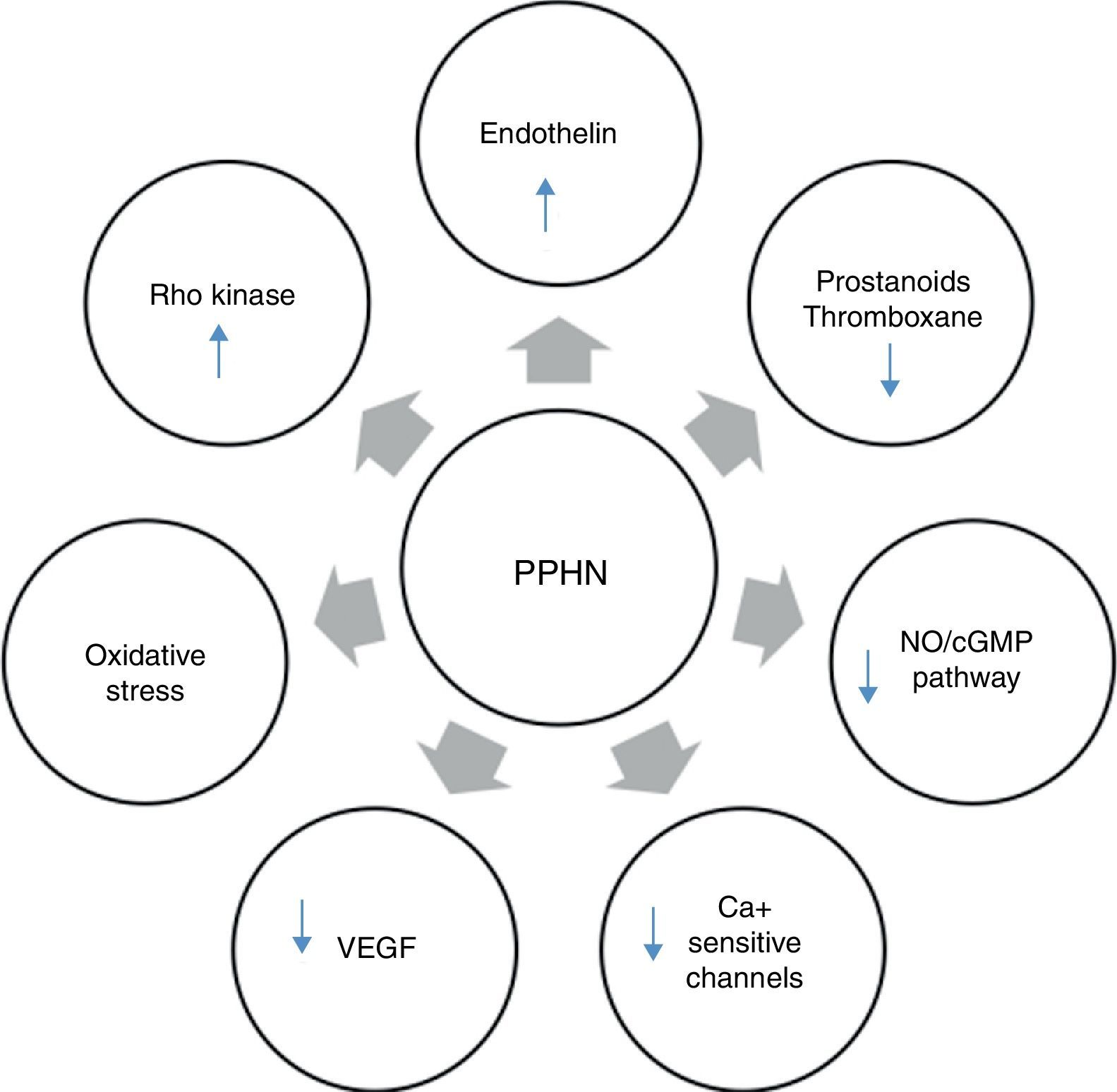
Neurohumoral mediators in persistent pulmonary hypertension of the newborn. Factors contributing to the physiologic alterations of PPHN include: (i) decreased production of vasodilator substances including NO and prostanoids; (ii) increase in endothelin production from the pulmonary endothelium; and (iii) exposure to high concentrations of supplemental oxygen leading to increased oxidative stress, activation of signaling pathways such as Rho kinase and alterations in expression of calcium-sensitive potassium channels. cGMP: cyclic guanosine monophosphate; NO: nitric oxide; PPHN: persistent pulmonary hypertension of the newborn; VEGF: vascular endothelial growth factor.
Meconium aspiration syndrome (MAS) is the most common cause of PPHN, affecting 25000–30000 infants with up to 1000 deaths annually in the United States.25 Aspiration occurs with the first breath after birth or, in more severe cases, in utero.26 Common disturbances of lung function in MAS include hypoxemia and diminished lung compliance. Low oxygenation is due to a combination of ventilation-perfusion mismatching, intrapulmonary shunting linked to regional atelectasis, and extrapulmonary shunting due to PPHN.27 Among the pathophysiologic mechanisms of hypoxemia in MAS are acute airway obstruction, dysfunction or inactivation of pulmonary surfactant (due to the presence of surfactant inhibitors such as albumin, phosphatidylserine, and phospholipase A2), chemical pneumonitis, consequent release of vasoconstrictor (ET-1, TXA2, and PGE2) and inflammatory mediators, and right-to-left extrapulmonary shunting involving PPHN.26 A few hours after meconium aspiration, neutrophils and macrophages are found in the alveoli and lung parenchyma. There is a release of cytokines, including tumor necrosis factor-alpha, interleukin 1-beta and interleukin-8, which can directly injure the lung parenchyma and lead to vascular leakage that causes pneumonitis with pulmonary edema.26
Idiopathic PPHN (IPPHN) is the second most common cause of PPHN and may be present in near-term and term newborns.10 Many cases of severe disease are associated with chronic intrauterine stress, severe hypertensive structural remodeling, vessel wall thickening and smooth muscle hyperplasia.10 Consequently, the pulmonary vasculature does not dilate properly in response to birth, inducing severe hypoxemia at birth. The pathophysiology of IPPHN is multifactorial; it may be due to constriction of the fetal ductus arteriosus from exposure to nonsteroidal anti-inflammatory drugs (NSAIDs), biologic or genetic susceptibility, or reactive oxygen species (ROS) such as superoxide and hydrogen peroxide.
In utero exposure to NSAIDs during the third trimester may lead to ductus arteriosus closure with a rapid increase in fetal pulmonary artery pressure, pulmonary vascular remodeling, and subsequent failure of neonatal transition. Inhibition of prostaglandin synthesis in neonates can lead to physiologic effects including disruption of the sleep cycle, increased danger of PH, cerebral blood flow changes, decreased renal function and disordered thermoregulation.28
PGI2 is important in the normal pulmonary vascular transition. Decreased expression of the prostacyclin synthase (PGIS) and the prostacyclin IP receptor has recently been demonstrated in PPHN lambs, leading to reduced vasodilation to prostacyclin analogues.10 PGI2 increases cAMP by stimulating adenylate cyclase in vascular smooth muscle, resulting in vasodilation. Aerosolized PGI2 has been shown to be at least as effective as iNO in decreasing PH in some animal models and humans. When combined with milrinone it reduces PVR by an additional 8% compared with PGI2 alone.29,30
Dysregulation of phosphodiesterase type 3 (PDE3) expression and activity can interfere with normal cAMP signaling in pulmonary vascular smooth muscle.10 While fetal levels of PDE3 are not altered by PPHN, PPHN suppresses the normal rise in PDE3 expression and activity following delivery and mechanical ventilation. Interestingly, NO increases PDE3 expression and activity in these lambs, demonstrating the interrelated nature of the cGMP and cAMP pathways in PPHN.30
Infants with PPHN have been shown to have decreased expression and activity of eNOS. Increased production of ROS due to various factors leads to vasoconstriction, smooth muscle proliferation, and eNOS dysfunction.31 PPHN is associated with severe hypoxemia, so the use of high oxygen concentrations (up to 100% oxygen) is typically considered as a first-line therapy in infants with PPHN.9 However, oxygen use can aggravate oxidative stress in multiple cellular compartments of the affected vasculature. Hyperoxia diminishes vascular responses to endogenous and exogenous NO in both normal and altered pulmonary vasculature, showing that ROS can inactivate NO or enzymes responsible for mediating its vascular effects. The underlying mechanisms by which ROS lead to pulmonary vasoconstriction and diminished NO responsiveness involve multiple targets in pulmonary vascular regulatory pathways, including PDEs. Exposing normal fetal pulmonary artery smooth muscle cells (FPASMC) to hyperoxia for 24 hours leads to decreased cGMP response to exogenous NO10 and increases phosphodiesterase type 5 (PDE5) mRNA and protein expression as well as increased levels of PDE5 phosphorylation and activity. It also increases oxidative stress within FPASMC. ROS may thus be a mediator between oxygen and PDE5. The changes in PDE5 expression and activity as well as the decreased cGMP responsiveness in hyperoxia are reversed with pretreatment with a chemical antioxidant, N-acetyl-cysteine. ROS, such as superoxide and hydrogen peroxide, are sufficient to increase PDE5 expression and activity in FPASMC that may promote vasoconstriction, poor NO response, and vascular remodeling.13
Ventilation with 100% oxygen significantly increases pulmonary vascular PDE5 expression and activity,10 representing that PDE5 activity is a significant moderator in the vascular response to oxygen and hyperoxia. PPHN lambs with 100% oxygen ventilation demonstrate pulmonary vasoconstriction and reduced pulmonary vascular relaxation in response to iNO. Recent studies show that an exaggerated increase in PDE5 activity to some extent mediates these abnormal reactions.10
ManagementPrimary treatment of the neonate with PH depends on the underlying disorder. Nevertheless, treatment in PPHN aims to reduce the size of the right-to-left shunt by controlling systemic blood pressure and administering pulmonary vasodilators (Table 2). For the majority of PPHN infants treatment frequently includes aggressive support of cardiac function and perfusion, with volume and inotropic agents to enhance cardiac output and systemic O2 transport.
Treatment objectives in persistent pulmonary hypertension of the newborn.
| 1. Correction of the underlying cause |
| 2. Ventilation support: decrease hypoxemia and hypercapnia |
| 3. Pulmonary vasodilators |
| 4. Correction of metabolic disorders: hypoglycemia, hypocalcemia and hypomagnesemia |
| 5. Nutritional support |
Ventilation is crucial for PPHN treatment since it facilitates alveolar recruitment and lung expansion, improving ventilation/perfusion matching.2
Oxygen is well known as a pulmonary vasodilator and should be started at 100%. Nevertheless, high oxygen concentrations should not be maintained for long periods, in order to prevent hyperoxia.32
High-frequency oscillatory ventilationIn severe cases, which do not respond to classical ventilation, high frequency oscillatory ventilation (HFOV) may have a place.33
As it involves very low tidal volumes, HFOV offers less barotrauma. It has been used mainly in cases of pulmonary hypoplasia (diaphragmatic hernia, pulmonary hypoplasia and severe MAS). However, there are insufficient data to determine its effect on mortality.19
Some studies suggest that iNO in combination with HFOV results in better oxygenation in patients with severe PPHN, and indicate that improved lung inflation during HFVO may enhance the response to iNO by reducing the intrapulmonary shunt and improving iNO distribution to the pulmonary circulation.26 Recent evidence shows that a small number of infants treated with HFOV and iNO will not respond and go on to extracorporeal membrane oxygenation (ECMO).27
Extracorporeal membrane oxygenationIndications for treatment with ECMO include34: (i) severe respiratory failure unresponsive to conventional treatment (FiO2=100%, hyperventilation, and drugs); (ii) birth weight over 2000 g; (iii) less than seven days of assisted ventilation; (iv) reversible pulmonary disease; (v) absence of congenital heart disease; and (vi) intraventricular hemorrhage or severe coagulopathy. Idiopathic PPHN patients have good survival with ECMO.35
VasodilatorsNitric oxideThe key to treating PPHN lies in providing selective pulmonary vasodilation.33 iNO is an appropriate option because NO is a fast and powerful vasodilator when delivered by inhalation therapy.31,36 iNO is also believed to have a ‘micro-selective effect’ that can improve ventilation/perfusion matching.36 Numerous trials involving the use of iNO in term and near-term infants with PPHN have demonstrated that it considerably decreases the need for ECMO in newborns with PPHN.31 Nevertheless, 40% of infants are non-responders to iNO.32 This indicates that the clinical problem of PPHN is more complex than a simple deficiency in NO production.36 However, iNO does not decrease the incidence of chronic lung disease or adverse neurodevelopmental sequelae.10 Contraindications to iNO include congenital heart disease with left ventricular outflow tract obstruction, critical aortic stenosis, hypoplastic left heart syndrome, and severe left ventricular dysfunction.36 When there is no initial response to iNO and for those who deteriorate during iNO therapy, alternative therapy may be required.
SildenafilSildenafil is a phosphodiesterase type 5 (PDE5) inhibitor that reduces pulmonary vascular resistance in both animal models and human adults and is an effective treatment for PPHN.10 Inhibition of hyperoxia-induced PDE5 activity with sildenafil partially rescues the cGMP response to exogenous NO, further indicating that PDE5 is a critical regulator of cGMP in the context of hyperoxia.10,32 The use of sildenafil in the treatment of pediatric pulmonary arterial hypertension (PAH) may increase long-term mortality.37
MilrinoneMilrinone is a positive inotrope and vasodilator; its mechanism of action involves increasing cAMP levels by inhibiting phosphodiesterase type 3 (PDE 3) in cardiac and vascular smooth muscle.30 Intravenous milrinone leads to better oxygenation and improvements in pulmonary and systemic hemodynamics in patients with suboptimal response to iNO.38 Recent studies have shown that milrinone, when used together with iNO or iloprost, can help to restore normal vascular responses.10 Pretreatment with milrinone restores relaxation to prostanoids to levels similar to that seen in control fetuses.30
BosentanBosentan, a dual ET receptor antagonist, is a well-established therapeutic option in adult PAH. Further investigation for its use in PPHN is needed.9 However, studies have concluded that bosentan is safe and effective for the treatment of PAH in children, with improved WHO functional class and significant falls in mean pulmonary artery pressure and PVR.39 A single trial showed that bosentan was more effective than placebo in improving oxygen inhalation and oxygen saturation and decreasing mechanical ventilation time.40,41
Support therapyPatients with PPHN usually need vasopressor support. Dopamine is the drug of choice when neonates require pharmacologic inotropic support.42
In patients with severe respiratory failure born at full term, the use of surfactant has been shown to decrease the need for ECMO.9,43
Acidosis also raises PVR and must be rectified. Nevertheless, alkali infusion to sustain alkalosis is not recommended,44 since prolonged alkalosis may paradoxically worsen pulmonary vascular tone, reactivity and permeability edema.1
PPHN patients can have patient-ventilator dyssynchrony, resulting in agitation, which causes catecholamine release and raising PVR. Therefore, in ventilated infants the use of an opioid analgesic can be useful. If dyssynchronous breathing and severe hypoxemia persist, and a specific cause cannot be identified, neuromuscular blockade can be used.44
New treatment approachesAdvances in the treatment of PPHN over the past decade have enabled physicians to substantially improve its prognosis, but mortality remains high.40 Current treatments are centered predominantly on vasodilation, while many emerging therapies are directed at cell proliferation and remodeling.
One mechanism that could be exploited to treat PPHN is to improve endogenous NO bioavailability by increasing the activity of eNOS. Cicletanine improves eNOS coupling, thereby increasing bioavailable NO.40
Another novel therapy involves endothelial progenitor cells (EPCs), which may play a role in the pathogenesis of the disease.45 Therapeutic administration of EPCs was shown to improve PH in animal models, with enhanced efficacy when EPCs were transduced with eNOS.26
Oxidant stress is known to contribute to the endothelial dysfunction associated with vascular disease. The enzyme responsible for the clearance of superoxide anion, superoxide dismutase, has been used clinically, including in premature infants. One study found that recombinant human superoxide dismutase reduced PVR and was synergistic with iNO in a fetal lamb model of PPHN patients.40
Vasoactive intestinal peptide (VIP) is a potent pulmonary and systemic vasodilator. An acute vasodilator response has been observed following the inhalation of VIP in patients with idiopathic PAH. Adrenomedullin and vasoactive intestinal peptide have been shown to be upregulated in PH and to reverse disease progression in animal models.45
Rho kinase inhibition targets pulmonary vascular smooth muscle. Increased RhoA and Rho kinase activities have been demonstrated in the lungs, platelets, and pulmonary artery smooth muscle cells harvested from patients with idiopathic PH. Therapy aimed at altering Rho kinase activity alteration has the potential advantage, as opposed to endothelial-based therapies, of directly affecting the contractile apparatus of the smooth muscle cell. Rho kinase inhibition has potential efficacy in PH treatment, but large controlled trials are needed (Table 3).40
ConclusionPPHN may be primary, or secondary to a variety of conditions such as intrapartum asphyxia, infection, pulmonary hypoplasia, congenital heart disease or drug therapy. It can be associated with a normal number (maladaptation) or a decreased number of arteries (as in pulmonary hypoplasia). Factors contributing to the physiologic alterations of PPHN include reduced production of vasodilator substances and increased production of vasoconstrictor substances by the pulmonary endothelium, increased oxidative stress and triggering of signaling pathways such Rho kinase. Few treatment strategies used in infants with PH of the newborn have been subjected to rigorous evaluation. Inhaled NO has been shown to reduce the need for extracorporeal membrane oxygenation but not mortality in term or near term infants. Current evidence indicates that different approaches may improve oxygenation (such as with sildenafil, bosentan or milrinone) and new vasodilators are available. Emerging therapies are directed at cell proliferation and remodeling, but long-term trials are necessary to assess these promising approaches.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that no patient data appear in this article.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflicts of interestThe authors have no conflicts of interest to declare.
