




Risk stratification of sudden death in patients with Brugada syndrome (BrS) is a controversial issue, and there is currently no consensus on the best method. Examination of data from the natural history of the disease is of fundamental importance and may help to identify relatives at risk. At the same time, study of the genetic mutations responsible for the disease may also contribute to risk stratification of the syndrome, enabling identification of asymptomatic relatives carrying mutations.
This paper presents the case of a young man, aged 26, monitored as a pediatric cardiology outpatient from birth for a simple structural heart defect not requiring surgery. Analysis of the evolution of the patient's electrocardiogram revealed the appearance, at the age of 20, of a pattern compatible with type I BrS. Following an episode of syncope and induction of polymorphic ventricular tachycardia in the electrophysiological study, a cardioverter-defibrillator was implanted. One year later, a single shock terminated an episode of ventricular fibrillation. A molecular study of the SCN5A gene identified a rare mutation, c.3622G>T (p.Glu1208X), recently described and associated with more severe phenotypes in patients with BrS, as in the case presented.
A estratificação do risco de morte súbita nos doentes com síndrome de Brugada (SB) é um assunto controverso, não existindo atualmente consenso sobre a forma ideal de o fazer. O estudo da história natural da doença é fundamental e pode ajudar a identificar os familiares em risco. Por outro lado, o estudo das mutações genéticas responsáveis pela síndrome pode contribuir para a estratificação do risco, identificando os familiares assintomáticos portadores de mutação.
Este artigo apresenta o caso de um jovem de 26 anos de idade, seguido na consulta de Cardiologia Pediátrica desde o nascimento por um defeito cardíaco estrutural simples, que resolveu espontaneamente. A análise evolucionária do eletrocardiograma do doente documentou o aparecimento, aos 20 anos de idade, de um padrão compatível com SB de tipo 1. Após um episódio de síncope e indução de taquicardia ventricular polimórfica no estudo electrofisiológico, foi implantado um cardioversor-desfibrilador. Um ano depois, um episódio de fibrilhação ventricular foi terminado por um choque único. O estudo molecular do gene SCN5A identificou uma mutação rara [c.3622G>(p.Glu1208X)], recentemente descrita e associada a fenótipos mais graves nos doentes com SB, tal como no caso por nós apresentado.
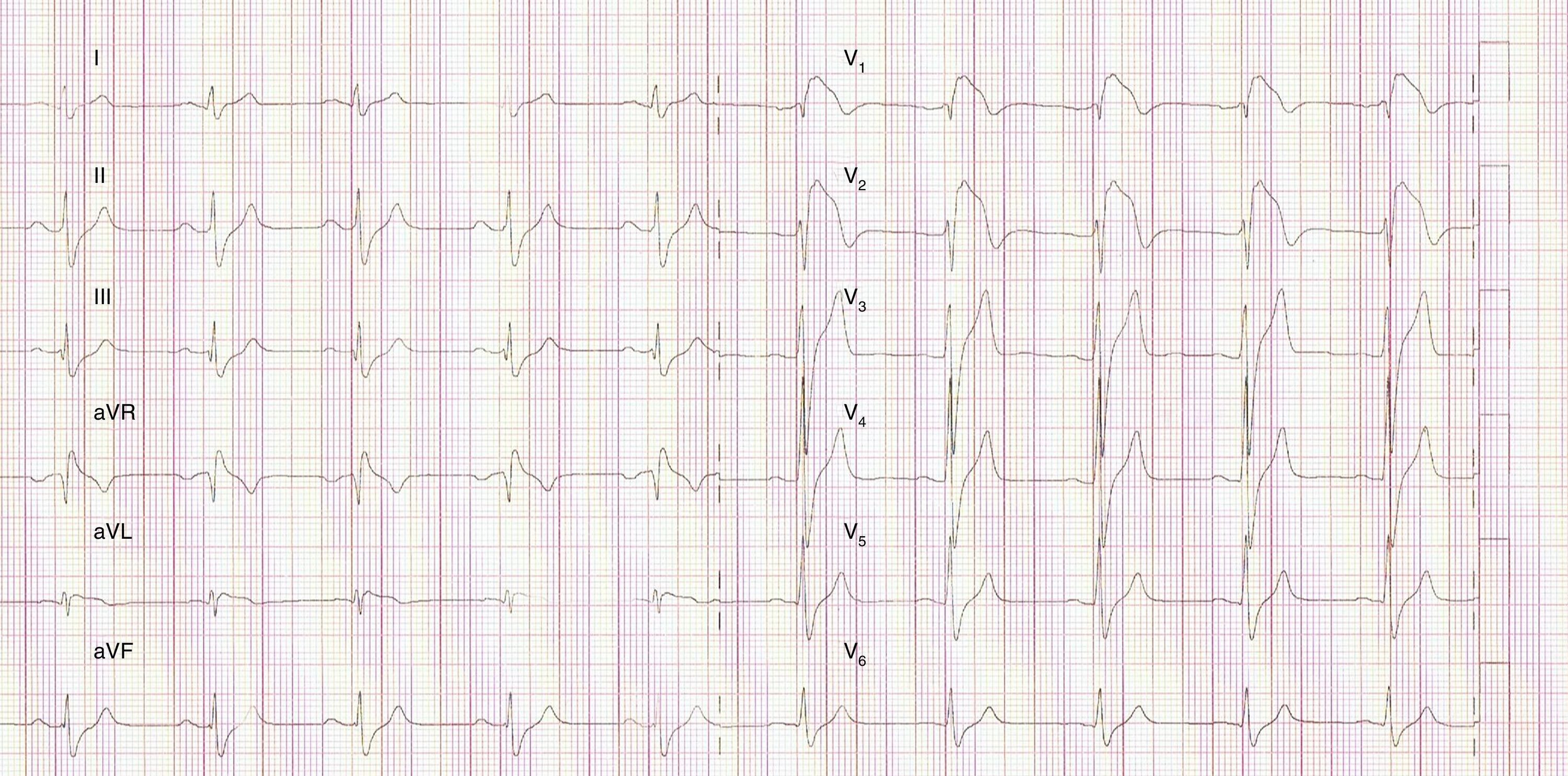
We present the case of a young man, aged 26, monitored as a pediatric cardiology outpatient from birth for a perimembranous ventricular septal defect. The patient's family history included sudden death of his maternal grandfather in the fifth decade of life, and “arrhythmia” in his father, controlled with propranolol. The initial electrocardiogram (ECG), at age one month, presented a predominance of R waves and ST depression in the right precordial leads (Figure 1). During follow-up, the ventricular septal defect was observed to become restrictive and eventually closed spontaneously. The patient progressed without complications until the age of 20, at which time the ECG pattern changed, with the appearance of complete right bundle branch block and ST-segment elevation in the right precordial leads (Figure 2), suggesting type I Brugada syndrome (BrS). The patient was monitored irregularly, due to his good health and failure to attend appointments. He appeared to remain asymptomatic until the age of 24, when he presented a first episode of syncope, at rest, during the night, not preceded by prodromal symptoms or fever and accompanied by tonic-clonic movements and sphincter incontinence. This episode was initially interpreted as a seizure, leading to a brief hospitalization. Following this episode and in view of the ECG features, an electrophysiological study was conducted, during which sustained polymorphic ventricular tachycardia was induced. A single-chamber implantable cardioverter-defibrillator (ICD) was implanted. Three months later, the patient suffered an episode of syncope followed by a shock. Interrogation of the device revealed that the shock was appropriate and due to ventricular fibrillation. The current monitoring period (subsequent to ICD implantation) has lasted for two years, with no new episodes of arrhythmia to date, and without drugs. The patient and his family were referred to the cardiogenetics clinic for genetic counseling prior to the decision to conduct the molecular study, and neuropsychological support was initiated. As well as genetic counseling, all family members underwent an ECG, with normal results. The patient's genetic study was conducted on the SCN5A gene, which is responsible for 15–30% of BrS mutations.1,2 A pathogenic mutation in heterozygosity, c.3622G>T (p.Glu1208X), and the polymorphisms c.87A>G (p.Ala29Ala) in homozygosity and c.3183A>G (p.Glu1061Glu) and c.5457T>C (p.Asp1819Asp) in heterozygosity were identified. The variant c.3841-24C>T was also identified, without clinical significance to date. Molecular study of first-degree relatives is currently under way.
Discussion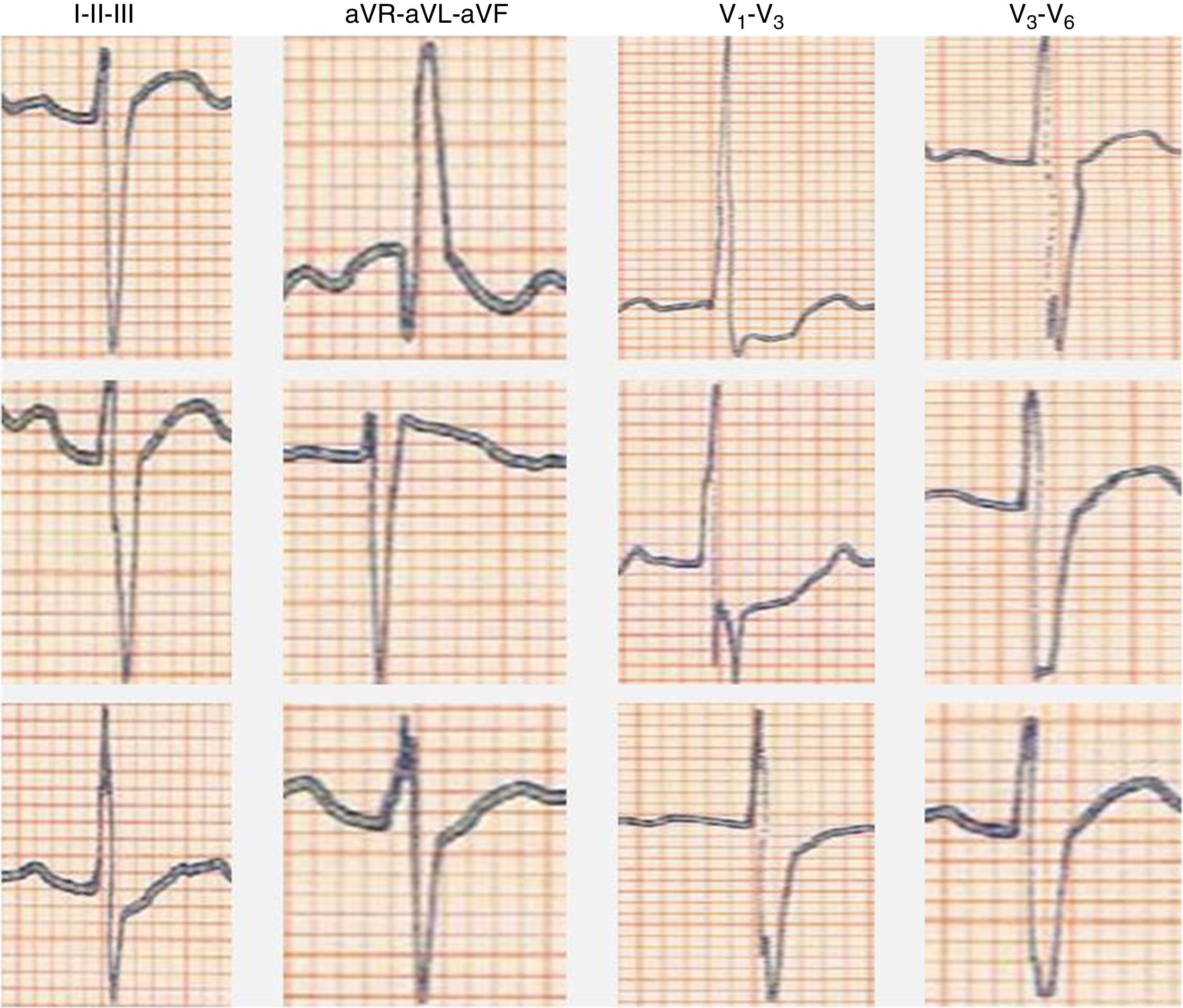
BrS is an autosomal dominant hereditary heart disease with incomplete penetrance and a mean prevalence of 5:10000 in Europe.3 The syndrome occurs more frequently in patients of Asian origin, in particular in Japan and south-east Asia, especially Thailand and the Philippines, where prevalence is estimated at up to 12:10000.4 BrS is characterized by the presence of electrocardiographic changes (incomplete right bundle branch block and ST-segment elevation in the right precordial leads) and a tendency for sudden cardiac death caused by polymorphic ventricular tachycardia or ventricular fibrillation.3 The ECG alterations may not be always present, surfacing as a result of fever, the use of sodium channel blockers, vagolytic agents, beta-blockers and others.3 BrS is responsible for over 4% of all sudden deaths and 20% of sudden deaths in patients with structurally normal hearts.3 It is more frequent among males, and the mean age for the first manifestations is 40, although it can occur at younger ages, as shown by the case under examination, or older ages.3 The risk of sudden death in these patients over a 24-month period has been estimated at 8%.3
Molecular studies conducted in the late 1990s demonstrated a relationship between mutations in genes which encode ion channels and the existence of hereditary lethal arrhythmias.5 In BrS, mutations in 10 genes have been identified.4 Mutations in the SCN5A gene, located in the 3p22 region, responsible for encoding sodium channels, are present in around 15–30% of patients clinically diagnosed with BrS.1,2,5 This gene is also responsible for type 3 long QT syndrome.6 Mutations in this gene can result, by different mechanisms, in a reduction of the function or number of sodium channels in myocardial cell membranes.6 The disease's penetrance and expressivity are highly variable, and thus different instruments for stratification of the syndrome's arrhythmic risk are needed. There is currently no consensus on the best way to achieve this goal. The following have been proposed as arrhythmic risk factors: spontaneous ST-segment elevation (type I), existence of a previous episode of syncope or aborted sudden death, documented (spontaneous) ventricular tachycardia, and inducibility of sustained ventricular tachycardia by programmed ventricular stimulation.4 In this regard, we highlight the importance of obtaining data pertaining to the natural history of the disease and family background of sudden death and arrhythmia, in the context of which a medical genetics, cardiology and arrhythmology team may help to identify relatives potentially at risk as well as asymptomatic mutation carriers.
In this case, regular evaluation of the patient's electrocardiogram and its changes over time led to diagnosis of the disease, subsequently confirmed by genetic study. The polymorphisms identified in the molecular study are already described non-pathogenic variants, although variant c.3841-24C>T has never been described. The significance of this variant for the BrS phenotype is unclear. The pathogenic mutation p.Glu1208X was recently described in a patient.7 It causes a premature stop codon (truncation mutation), leading to a 100% reduction in sodium channel current.
In all series published to date, there have been no differences in terms of arrhythmic events when patients are divided according to the presence or absence of SCN5A mutations.5 Meragalli et al.7 proposed, for the first time, that mutations causing a drastic reduction in sodium channel activity result in more severe phenotypes, with a greater number of syncopes and longer PR intervals on the ECG. It is possible that, in the future, molecular study of SCN5A mutations will enable identification of high-risk BrS patients. The existence of common polymorphisms on the same gene may modulate the effect of BrS mutations,8,9 modifying the phenotype. Such polymorphisms could in the future be the target of new therapeutic interventions.
We conclude that knowledge of the natural history of the disease and of the inter-relationship between phenotype and genotype are necessary to guide stratification of arrhythmic risk in BrS. Clinical follow-up and treatment, along with customized predictive and preventive intervention for patients and relatives, should be conducted within the scope of a multidisciplinary team covering the fields of pediatric cardiology, cardiology, arrhythmology, clinical psychology and medical genetics.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
Conflicts of interestThe authors have no conflicts of interest to declare.
The authors acknowledge the assistance of Instituto de Medicina Molecular.
