








As estatinas são os fármacos mais prescritos no tratamento da dislipidemia, estando recomendadas na prevenção primária e secundária das doenças cardiovasculares. Para além de diminuírem a síntese de colesterol, interferem com a síntese de intermediários isoprenoides, o que poderá explicar muitos dos seus efeitos pleiotrópicos, incluindo a ação antioxidante.
O stresse oxidativo é definido como um desequilíbrio entre a formação de espécies reativas de oxigénio e a sua eliminação por sistemas de defesa antioxidantes, com prevalência de um estado pró‐oxidante com efeitos deletérios nas macromoléculas orgânicas e na sinalização redox celular. As espécies reativas de oxigénio podem interferir com vários processos que afetam a estrutura e função cardíacas, contribuindo para a disfunção contráctil, fibrose e hipertrofia do miocárdio observadas na fisiopatologia da insuficiência cardíaca. As estatinas promovem a restauração do equilíbrio redox pela regulação de várias vias moleculares responsáveis pelo controlo da atividade de enzimas como a NADPH oxídase e a sintetase endotelial de monóxido de azoto. Estes fármacos contribuem também para o controlo de processos inflamatórios e parecem desempenhar um papel protetor em várias patologias. Os resultados de estudos observacionais e ensaios clínicos, realizados com o objetivo de esclarecer o efeito das estatinas na insuficiência cardíaca, não têm sido consensuais. Esta revisão tem como principal objetivo analisar o papel do stresse oxidativo na insuficiência cardíaca e os mecanismos moleculares inerentes às propriedades antioxidantes das estatinas. Pretende ainda reunir a evidência científica atual relativa à utilização destes fármacos como terapêutica específica da insuficiência cardíaca.
Statins are the most commonly prescribed drugs for the treatment of dyslipidemia. They are also recommended in primary and secondary prevention of cardiovascular disease. In addition to decreasing cholesterol synthesis, statins interfere with the synthesis of isoprenoid intermediates, which may explain many of their pleiotropic properties, including their antioxidant effects.
Oxidative stress is defined as an imbalance between the synthesis of reactive oxygen species and their elimination by antioxidant defense systems, with a prevailing pro‐oxidant status that results in macromolecular damage and disruption of cellular redox signaling. Reactive oxygen species interfere with various processes that affect cardiac structure and function, contributing to the contractile dysfunction, myocardial hypertrophy and fibrosis observed in the pathophysiology of heart failure. By regulating several molecular pathways that control nicotinamide adenine dinucleotide phosphate oxidase and endothelial nitric oxide synthase activity, statins help restore redox homeostasis. These drugs also contribute to the control of inflammation and appear to have a protective role in various diseases. The results of observational studies and clinical trials with statins in heart failure have not been consensual.
This review aims to analyze the role of oxidative stress in heart failure and the molecular mechanisms underlying statins’ antioxidant properties. It also examines current scientific evidence on the use of these drugs as a specific treatment for heart failure.
Colégio Americano de Cardiologia
Ácido desoxirribonucleico
Cínase B de proteínas
Cínase de proteínas ativada pelo monofosfato de adenosina
Proteína ativadora 1
Ácido ribonucleico mensageiro
Cínase reguladora da sinalização apoptótica, de tipo 1
Acidente vascular cerebral
Tetra‐hidrobiopterina
Peptídeo natriurético de tipo B
Bomba membranar de cálcio do retículo sarcoplasmático
Ensaio clínico multinacional controlado de avaliação da rosuvastina na Insuficiência Cardíaca
Ciclooxigenase 2
Superóxido dismútase–Cobre, Zinco (isoforma citoplasmática; SOD‐1)
Doença coronária
Doenças cardiovasculares
Proteína de tipo 2 de ligação a danos no ADN
Enfarte agudo do miocárdio
Superóxido dismútase extracelular (SOD‐3)
Sintetase endotelial de monóxido de azoto
Cínases reguladas por sinais extracelulares
Sociedade Europeia de Cardiologia
Fibrilhação auricular
Fração de ejeção
Fração de ejeção do ventrículo esquerdo
Farnesilpirofosfato
Grupo Italiano para o Estudo da Sobrevivência no Enfarte de Miocárdio–Insuficiência cardíaca
Geranilgeranilpirofosfato
Glutationa peroxídase
Glutationa (forma reduzida)
Trifosfato de guanosina
Ciclo‐hidrolase do GTP
Ácido hipocloroso
Lipoproteína de elevada densidade
Hidroximetilglutaril coenzima A
Heme oxigenase 1
Radical hidroxilo
Peróxido de hidrogénio
Insuficiência cardíaca
Lipoproteínas de baixa densidade
Monoaminoxídase
Cínases de proteínas ativadas por mitogénios
Malonildialdeído
Metaloproteases da matriz extracelular
Superóxido dismútase mitocondrial (SOD‐2)
Mieloperoxídase
Fator nuclear kappa B
Forma reduzida do dinucleotídeo fosfatado de nicotinamida e adenina
Associação Nova Iorquina do Coração
Monóxido de azoto
NADPH oxídase
N–terminal do proBNP
LDL oxidadas
Oxigénio
Superóxido
Cínase do 3‐fosfatidilinositol
Cínase A de proteínas
Paraoxonase‐1
Espécies reativas de azoto
Espécies reativas de oxigénio
Serina 1177
Serina 633
Serina 473
Forma solúvel da gp91phox
Superóxido dísmutase
Fator de necrose tumoral α
Xantina oxídase
5‐lipoxigenase
8‐hidroxi‐2′‐desoxiguanosina (8‐OHdG)
15‐epi‐lipoxina A4
A prevalência de insuficiência cardíaca (IC) tende a aumentar no mundo ocidental em cerca de 46% até 20301 e a sua taxa de mortalidade mantém‐se em redor dos 50% aos cinco anos após o diagnóstico2. Impõe‐se, portanto, a exploração das principais vias fisiopatológicas e rediscussão da terapêutica.
A IC é definida como uma síndrome clínica complexa, resultante de alterações estruturais ou funcionais que modificam a capacidade de enchimento ou de ejeção ventricular3, e na qual há evidência de desequilíbrios no estado redox e na produção de monóxido de azoto (NO) e que a terapêutica com benefício clínico tende a restabelecer4.
Em Portugal, as estatinas, inibidores da redutase da hidroximetilglutaril‐Coenzima A (HMG‐CoA) (Figura 1), correspondem a 90% do consumo de antidislipidémicos5, desconhecendo‐se, todavia, a prescrição na IC. Apesar de vários estudos observacionais sugerirem um possível benefício na mortalidade e morbilidade associadas à IC, em parte atribuído à sua ação antioxidante6–8, e de a doença coronária (DC) ser responsável por aproximadamente 2/3 dos casos de IC sistólica3 em que está bem estabelecida a importância desta terapêutica, a prescrição de estatinas na IC por si só tem sido colocada em causa pelos resultados obtidos em ensaios clínicos aleatorizados. O uso destes fármacos tem sido também limitado pela caquexia cardíaca e pelo efeito paradoxal de concentrações séricas baixas de colesterol se associarem a pior prognóstico na IC9.

Estrutura química dos inibidores da redutase HMG‐CoA. Adaptado de94.
A lovastatina, a sinvastatina e a pravastatina são de origem fúngica, enquanto as estatinas mais recentes são de origem sintética. Os fármacos de origem fúngica são estruturalmente semelhantes e possuem um anel hidronaftaleno em comum. Enquanto a sinvastatina e a lovastatina são administrados como profármacos inativos, a pravastatina é administrada na sua forma ativa. As restantes estatinas de origem sintética apresentam estruturas distintas que influenciam a sua solubilidade em meio aquoso.
O principal mecanismo de ação das estatinas resulta da inibição da redutase da HMG‐CoA, a enzima limitante da síntese de colesterol. O bloqueio desta enzima interfere com a síntese do mevalonato e de intermediários isoprenoides, nomeadamente, o farnesilpirofosfato (FPF) e o geranilgeranilpirofosfato (GGPF), produtos responsáveis pela isoprenilação de uma grande variedade de proteínas entre as quais as pequenas proteínas associadas ao trifosfato de guanosina (GTP) como a Ras, Rho e Rac (Figura 2). A isoprenilação destas moléculas é fundamental para a formação de ligações covalentes, definição da localização subcelular e translocação intracelular de proteínas associadas à membrana10. É possível que a inibição da síntese dos intermediários isoprenóides seja responsável por vários dos efeitos pleiotrópicos das estatinas (Tabela 1)11, tais como as ações antioxidantes e anti‐inflamatórias.
 eNOS – sintetase endotelial do monóxido de azoto; ET‐1 – endotelina ‐1; HMG‐CoA – hidroximetilglutaril Coenzima A; PAI‐1 – inibidor do ativador do plasminogénio tipo 1; PP – pirofosfato; Rac1 e RhoA – proteínas G de baixo peso molecular; t‐PA – ativador do plasminogénio tecidual.' title='Efeito das estatinas na via do mevalonato. Adaptado de95,96. ↑ – aumento;↓ – diminuição;
eNOS – sintetase endotelial do monóxido de azoto; ET‐1 – endotelina ‐1; HMG‐CoA – hidroximetilglutaril Coenzima A; PAI‐1 – inibidor do ativador do plasminogénio tipo 1; PP – pirofosfato; Rac1 e RhoA – proteínas G de baixo peso molecular; t‐PA – ativador do plasminogénio tecidual.' title='Efeito das estatinas na via do mevalonato. Adaptado de95,96. ↑ – aumento;↓ – diminuição; Efeito das estatinas na via do mevalonato. Adaptado de95,96.
↑ – aumento;↓ – diminuição; eNOS – sintetase endotelial do monóxido de azoto; ET‐1 – endotelina ‐1; HMG‐CoA – hidroximetilglutaril Coenzima A; PAI‐1 – inibidor do ativador do plasminogénio tipo 1; PP – pirofosfato; Rac1 e RhoA – proteínas G de baixo peso molecular; t‐PA – ativador do plasminogénio tecidual.
Efeitos pleiotrópicos das estatinas e vias moleculares associadas
| Propriedades das estatinas | Mecanismos de ação |
|---|---|
| Anti‐inflamatória | ↓ MPO (enzima pró‐oxidante secretada por neutrófilos e monócitos em condições inflamatórias)69↑ 15‐epi‐lipoxina A4 (mediador eicosanoide da resolução da inflamação)63↑ prostaciclina vasodilatadora e anti‐inflamatória através da ativação da via da PLA2‐COX97↑ fator de transcrição KLF2 nas células endoteliais97↓ número de células inflamatórias nas placas ateroscleróticas através da redução de moléculas de adesão celular como a ICAM‐110 |
| Antioxidante | Inibição da Nox2 plaquetária75↓ concentração circulante da subunidade catalítica da Nox247↑ adiponectina (inibidor da ativação da Nox)47↑ catálase e ↑ SOD através da fosforilação da cínase de proteínas Akt41↑ transcrito, expressão proteica e ↑ atividade enzimática da SOD através da repressão do ADN da proteína DDB2 (regulador epigenético negativo da transcrição do gene da SOD)48↑ atividade da catálase e ↑ glutationa48↑ atividade da enzima antioxidante da tioredoxina‐1 devido à S‐nitrosilação da enzima27↑ ARNm e expressão proteica da HO‐150↑ atividade da PON1 (enzima responsável pelas propriedades antioxidantes da lipoproteína de elevada densidade [HDL])51 |
| Melhoria função endotelial | ↑ ARNm da eNOS, a enzima responsável pela formação endotelial de NO39,40↑ do cofator (tetrahidrobiopterina) da eNOS39,40↑ da GTPCH, a enzima limitante da síntese de tetrahidrobiopterina (cofatorda eNOS)39,40↑ atividade da eNOS após fosforilação mediada pela via das cínases PI3K/Akt e PKA39↑ da estabilidade do ARNm da eNOS devido a poliadenilação42↑ atividade da eNOS após fosforilação da cínase AMPK44↓ caveolina‐1 (domínio lipídico que interage com a eNOS influenciando negativamente a sua atividade enzimática)45,46Inibição da expressão de endotelina‐1 (potente vasoconstritor e mitogénio)10 |
| Antiarrítmica | Inibição da Nox79 |
| Angiogénica | ↑ proliferação, migração e sobrevida das células precursoras endoteliais10↑ mobilização das células precursoras endoteliais da medula óssea e aceleração da formação da estrutura vascular via PI3K/Akt e eNOS10 |
| Antitrombótica e antiplaquetária | Inibição da Nox2 plaquetária75↑ fator de transcrição KLF297↑ eNOS, ↓ TXA2 e modificação do conteúdo de colesterol nas membranas plaquetárias10 |
| Estabilização das placas ateroscleróticas | ↓ tamanho e modificação das propriedades físico‐químicas da porção lipídica da placa aterosclerótica10↓ acumulação macrófagos e ↓ produção de enzimas proteolíticas, as metaloproteases10 |
| Prevenção da proliferação das células musculares lisas | Bloqueio do ciclo celular entre a transição das fases G1/S10Inibição da pequena GTPase RhoA mediadora da proliferação das CML induzida pelo fator de crescimento derivado das plaquetas (PDGF) 10 |
| Prevenção da hipertrofia e remodelação cardíaca | ↓ fosforilação das cínases Akt e ERK1/2 e do fator de transcrição GATA498↑ expressão do ARNm do PPARα, um fator de transcrição membro da família de recetores nucleares ativados por ligandos98 |
| Imunomoduladora | ↓ maturação das células apresentadoras de antigénio através da ↓ das moléculas do complexo de histocompatibilidade classe II99Inibição da captação de antigénios pelas células apresentadoras de antigénios99Inibição da secreção de citocinas através da supressão do fator de transcrição NF‐KB99 |
| Modulação do SNA | ↓ noradrenalina e ↓ atividade simpática renal100↓ ARNm e da expressão proteica do recetor da angiotensina e das subunidades da Nox100↓ superóxido no bolbo raquidiano ventrolateral rostral100 |
↑ – aumento; ↓ – diminuição; ARNAm – Ácido ribonucleico mensageiro; Akt – cínase B de proteínas; AMPK – cínase de proteínas ativada pelo monofosfato de adenosina; CML – células musculares lisas; COX – ciclooxigenase; DDB2 – Proteína de tipo 2 de ligação a danos no ADN; eNOS – sintetase endotelial do NO; ERK1/2 – cínases 1 ou 2 reguladas por sinais extracelulares; GATA4 – proteína 4 de ligação ao GATA; GTPCH – ciclo‐hidrolase do trifosfato de guanosina; HO‐1 – heme oxigenase 1; ICAM‐1 – molécula de adesão intercelular‐1; KLF2 – fator Kruppel‐like 2; MPO – mieloperoxídase; NF‐KB – fator nuclear kappa B; NO – monóxido de azoto; Nox – NADPH oxídase, PDGF – fator de crescimento derivado das plaquetas; PI3K – cínase do 3‐fosfatidilinositol; PKA – cínase A de proteínas; PLA2 –fosfolípase A2; PON1 – paraoxonase 1; PPARα – recetor de tipo α ativado por proliferadores de peroxissomas, RhoA – proteína G de baixo peso molecular; SOD – superóxido dismútase; SNA – sistema nervoso autónomo; TXA2 – tromboxano A2.
Esta revisão tem como principal objetivo analisar o papel do stresse oxidativo na IC e os mecanismos moleculares inerentes às propriedades antioxidantes das estatinas. Pretende ainda reunir a evidência científica relativa à sua utilização como terapêutica específica da IC.
Insuficiência cardíaca e stresse oxidativoNos últimos anos, vários estudos experimentais e clínicos evidenciaram a importância do stresse oxidativo na patogénese da IC. O stresse oxidativo é definido como um desequilíbrio entre a formação de espécies reativas de oxigénio (ROS) e a sua eliminação por defesas antioxidantes, com prevalência de um estado pró‐oxidante que pode danificar macromoléculas orgânicas e desregular a sinalização redox celular12. A família das ROS inclui os radicais livres, como o superóxido (O2•−) e o radical hidroxilo (HO•), e as espécies oxidantes não radicais, como o peróxido de hidrogénio (H2O2) e o ácido hipocloroso (HClO)13.
Os cardiomiócitos, as células endoteliais e os neutrófilos produzem ROS no coração14. As principais fontes enzimáticas de ROS nestas células incluem as oxídases mitocondriais, as NADPH oxídases (Nox), a xantina oxídase (XO), a sintetase endotelial do NO (eNOS), as monoaminoxídases (MAO) e a mieloperoxídase (MPO)14–16.
As mitocôndrias são fontes primárias de ROS. Cerca de 90% do oxigénio (O2) é utilizado pelas oxídases mitocondriais para a produção de trifosfato de adenosina num processo acoplado à redução de O2 a água. Cerca de 1‐4% do oxigénio usado nestas reações é convertido em O2•− e H2O2, que podem ser prejudiciais para a função mitocondrial se não forem adequadamente neutralizados13. Os cardiomiócitos possuem a maior densidade de mitocôndrias do organismo, de modo a assegurar as necessidades energéticas. As mitocôndrias cardíacas provenientes de animais com IC produzem mais O2•− do que as mitocôndrias normais14.
As Nox são complexos enzimáticos que catalisam a redução do O2, usando a forma reduzida do dinucleotídeo fosfatado de nicotinamida e adenina (NADPH) como dador de eletrões. Neste processo são produzidas ROS, como o O2•− e o H2O217. Estas enzimas foram inicialmente caracterizadas nos neutrófilos. A estrutura das Nox nestas células inclui um complexo membranar formado pela subunidade catalítica gp91phox e por uma subunidade p22phox, as proteínas citoplasmáticas p47phox, p67phox e p40phox, e a Rac, uma proteína G de baixo peso molecular17,18. Foram já identificados vários homólogos da subunidade gp91phox, agora designada por Nox2. As Nox possuem assim sete isoformas, as Nox1‐5 e as oxídases duais, a Duox1 e Duox2, que diferem na expressão, composição molecular, localização subcelular e distribuição tecidual17,19. As Nox1‐3 requerem a translocação de subunidades citosólicas e a sua associação ao complexo membranar para a produção de ROS17. Já a Nox4 encontra‐se ativa constitutivamente e a Nox5 permanece num estado latente até que ocorra a estimulação pelo cálcio11,17. As Nox mais importantes no contexto da patologia cardiovascular são a Nox1, Nox2, Nox4 e Nox520. É de salientar que a atividade da Nox1 e da Nox2 pode ser estimulada pela angiotensina II, hormonas de crescimento e citocinas11,17. Quanto à distribuição, sabe‐se que as células endoteliais expressam Nox2, Nox4 e Nox5, enquanto as células musculares lisas vasculares expressam a Nox1, Nox4 e Nox5. Por sua vez, os fagócitos possuem sobretudo a Nox211. Os cardiomiócitos expressam principalmente as Nox2 e Nox419. A família das Nox tem sido implicada na patogénese da IC por sobrecarga de pressão, na IC induzida pela doxorrubicina e nas cardiomiopatias isquémica e diabética19.
A eNOS sintetiza NO e citrulina a partir do O2 e da L‐arginina. No entanto, em condições de stresse oxidativo ou de diminuição da disponibilidade do substrato (L‐arginina) ou do cofator (tetra‐hidrobiopterina, BH4), a eNOS pode ficar estruturalmente instável (desacoplada), passando a produzir O2•− em vez de NO21(Figura 3). No coração, a eNOS é expressa em células endoteliais e cardiomiócitos. O desacoplamento da eNOS parece contribuir para a disfunção endotelial na IC diastólica de etiologia hipertensiva e para a hipertrofia ventricular em resposta à sobrecarga de pressão14,22.
 eNOS. A síntese de NO pela
eNOS. A síntese de NO pela Efeito do stresse oxidativo na eNOS.
A síntese de NO pela eNOS requer a ligação de um cofator, a BH4. Quando a BH4 está ligada à eNOS, considera‐se que a enzima está acoplada. A degradação da BH4 pelas ROS, sobretudo pelo ONOO‐, determina o desacoplamento da enzima e, consequentemente, a produção de O2•− em detrimento do NO. Por sua vez, o O2•− pode reagir com o NO, originando ONOO− que induz a oxidação da BH4, promovendo assim um ciclo vicioso de desacoplamento da eNOS.
Akt – cínase B de proteínas; AMPK – cínase de proteínas ativada pelo monofosfato de adenosina; ADN – ácido desoxirribonucleico; ARNm – ácido ribonucleico mensageiro; BH4 – tetra‐hidrobiopterina; Cadeia PoliA – cadeia poliadenilada; Cav‐1 – caveolina‐1; eNOS – sintetase endotelial do monóxido de azoto; GTPCH‐1 – ciclohidrolase I do trifosfato de guanosina; NO – monóxido de azoto; O2 – oxigénio; O2•− – superóxido; ONOO− – peroxinitrito; P – fosfato; PI3K – cínase do 3‐fosfatidilinositol; PKA –cínase A de proteínas; ROS – espécies reativas de oxigénio; Ser1177 – serina1177da eNOS; Ser633 – serina633da eNOS.
A XO participa no metabolismo das purinas, catalisando a conversão da hipoxantina em xantina e desta em ácido úrico. Nestas reações, a XO utiliza o O2 como dador de eletrões originando O2•− e H2O213. O aumento da expressão e atividade da XO foi já descrito na IC. Por outro lado, o tratamento com alopurinol, um inibidor da XO, aumentou a contractilidade cardíaca em animais com IC e reduziu a remodelação cardíaca adversa num modelo experimental de enfarte do miocárdio14.
As MAO catalisam a degradação oxidativa de neurotransmissores, como a noradrenalina, a adrenalina e a dopamina, num processo que origina H2O2. Quando o coração está sujeito a stresse neuro‐hormonal e/ou hemodinâmico crónico, a elevada quantidade de monoaminas circulantes ou teciduais pode contribuir para o aumento da produção de H2O2 dependente das MAO15, com contribuições relevantes de cada isoforma. A MAO‐A poderá estabelecer uma ponte com o sistema renina‐angiotensina na fisiopatologia da cardiomiopatia diabética23, com evidência de necrose de cardiomiócitos quando sobre‐expressa24, enquanto a MAO‐B será preponderante em condições de sobrecarga hemodinâmica crónica25.
A MPO é uma enzima secretada por neutrófilos ativados e monócitos em condições inflamatórias, e utiliza o H2O2 para produzir várias espécies oxidantes (e.g. HClO, cloraminas, dióxidos de azoto) que podem causar lesões no coração e vasos sanguíneos e contribuir para a disfunção endotelial13. Em doentes com IC crónica verificou‐se o aumento da concentração plasmática da MPO em relação aos indivíduos controlo e a elevação da atividade desta enzima na IC crónica grave, comparativamente com o observado na IC ligeira a moderada16,26.
O stresse oxidativo pode resultar não só do aumento da síntese de ROS, mas também da disfunção das defesas antioxidantes13. As principais enzimas antioxidantes são a superóxido dismutase (SOD), a catálase e a glutationa peroxídase (GPx)13. A conversão do O2•− em H2O2 é catalisada pelas isoformas da SOD presentes no citoplasma e organelos (Cu,Zn‐SOD ou SOD‐1), nas mitocôndrias (MnSOD ou SOD‐2) ou no meio extracelular (EC‐SOD ou SOD‐3). O H2O2 pode depois ser convertido em H2O e O2 pela catálase presente nos peroxissomas ou pelas GPx presentes no citoplasma e mitocôndrias13. Existem ainda outras enzimas antioxidantes como, por exemplo, a glutationa redútase, a glutationa‐S‐transférase, as peroxirredoxinas e o sistema das tiorredoxinas. Estas últimas regulam o estado tiol/dissulfureto das proteínas, influenciando a sua estrutura e função. Adicionalmente, a tiorredoxina‐1 pode ligar‐se diretamente a ROS e metabolizá‐las27. A heme oxigenase 1 (HO‐1) é também uma enzima com ação antioxidante. Como a formação de ROS pode ser catalisada pelo excesso de heme, a degradação deste composto pela HO‐1 atenua o stresse oxidativo28. O nosso organismo possui ainda defesas antioxidantes não enzimáticas, como a glutationa (GSH), as vitaminas C e E, os carotenoides, o ácido úrico, a bilirrubina e a albumina13. A lipoproteína de elevada densidade (HDL) é outro exemplo de molécula antioxidante. Na HDL funcional há várias proteínas e enzimas com ação antioxidante e anti‐inflamatória, destacando‐se a enzima paraoxonase‐1 (PON‐1) que parece estar envolvida no seu efeito inibitório da oxidação das lipoproteínas de baixa densidade (LDL)29.
Apesar da diminuição das enzimas antioxidantes ter sido evidenciada em modelos animais30, em doentes com IC verificam‐se resultados contraditórios, estando descritos o aumento, a diminuição ou a ausência de alterações na atividade de enzimas antioxidantes31–33. Tem sido também observada a redução da vitamina C e de carotenoides sistémicos em doentes com IC crónica34.
As ROS podem interferir em vários processos que afetam a função e estrutura cardíacas, contribuindo para a génese e progressão da IC:
- •
ROS e contractilidade cardíaca
No coração, as ROS podem alterar a função de vários canais iónicos (canais de cálcio de tipo L, de sódio e de potássio) e inibir a atividade da bomba membranar de cálcio do retículo sarcoplasmático (Ca2+ ATPase SERCA2)35. Podem ainda induzir modificações de proteínas importantes para a contractilidade cardíaca. A fosforilação da troponina T por cínases ativadas por ROS pode contribuir para a redução da contractilidade cardíaca35.
- •
ROS, hipertrofia cardíaca e fibrose
As ROS estimulam várias enzimas da família das cínases de proteínas ativadas por mitogénios (MAPK), tais como as cínases reguladas por sinais extracelulares (ERK 1/2) e a cínase reguladora da sinalização apoptótica, de tipo 1 (ASK‐1), contribuindo para o desenvolvimento de hipertrofia cardíaca35. A ativação de fatores de transcrição como o fator nuclear‐κB (NF‐κB) e a proteína ativadora 1 (AP‐1) está também envolvida na hipertrofia cardíaca induzida pelas ROS35. As ROS podem ainda estimular a proliferação de fibroblastos cardíacos e ativar metaloproteases da matriz extracelular (MMP), com consequente remodelação da matriz extracelular. Estas alterações são importantes determinantes da remodelação miocárdica adversa14.
- •
ROS e apoptose dos cardiomiócitos
As ROS podem contribuir para a apoptose dos cardiomiócitos por vários mecanismos, incluindo genotoxicidade direta, ativação da cínase ASK‐1 em resposta ao fator de necrose tumoral‐α (TNF‐α) e estimulação de cínases indutoras do mecanismo de morte mitocondrial, em resposta à ativação de recetores adrenérgicos β35. É de salientar que a indução de hipertrofia ou apoptose nos cardiomiócitos depende da concentração de ROS. Assim, concentrações relativamente baixas de H2O2 induzem a síntese proteica, enquanto concentrações mais elevadas são responsáveis pela indução de apoptose nos cardiomiócitos35.
- •
ROS e disfunção mitocondrial
As mitocôndrias são importantes fontes de ROS no coração, mas podem também ser alvo dos seus efeitos tóxicos. As ROS podem danificar o ácido desoxirribonucleico (ADN) mitocondrial, com consequente diminuição de transcritos e da síntese proteica. Estes efeitos contribuem para o declínio da função mitocondrial, perturbação do metabolismo energético cardíaco e morte celular14.
- •
ROS e cardiomiopatia isquémica
As ROS podem contribuir para a génese e progressão da DC. As ROS produzidas na parede vascular estão envolvidas na formação do LDL oxidado (oxLDL) que desempenha um papel fundamental na patogénese da aterosclerose35. A ativação de MMP pelas ROS pode também contribuir para a instabilidade e rutura da placa de ateroma nas artérias coronárias e consequente trombose35. As ROS participam ainda na lesão de reperfusão e necrose tecidual, causadas pelo enfarte do miocárdio35.
- •
Interação entre ROS e espécies reativas de azoto (RNS)
A interação das ROS com o NO pode influenciar a função cardíaca. Sabe‐se que o NO medeia a S‐nitrosilação de proteínas em resíduos específicos de cisteína e que este processo afeta o fluxo de cálcio e o acoplamento excitação‐contração. Concentrações elevadas de O2•− podem inibir a S‐nitrosilação de proteínas, afetando o funcionamento cardíaco. Por outro lado, a reação do O2•− com o NO, além de contribuir para a disfunção endotelial por diminuição da disponibilidade de NO, origina também peroxinitrito, um potente oxidante que pode induzir apoptose ou necrose celular35,36.
Estatinas e stresse oxidativoEfeito das estatinas na eNOSA eNOS é a principal fonte enzimática de NO nos vasos sanguíneos. O NO contribui para a homeostasia vascular, inibindo a ativação e agregação plaquetárias, proliferação de células musculares lisas vasculares, expressão de moléculas de adesão celular e produção de matriz extracelular37. O NO regula também a contractilidade e frequência cardíacas, limita a remodelação cardíaca após o enfarte e contribui para o efeito protetor do pré e pós‐condicionamento isquémico38.
Como foi já referido, para que a eNOS sintetize NO é fundamental a ligação de BH4. A degradação deste cofator pelas ROS leva ao desacoplamento enzimático e, consequentemente, à produção de O2•− em detrimento do NO11 (Figura 3). A síntese de novo de BH4 envolve a enzima ciclo‐hidrolase I do GTP (GTPCH), que é um fator limitante para a formação de BH4. Em células endoteliais humanas, os tratamentos com fluvastatina ou cerivastatina aumentaram significativamente a expressão do ácido ribonucleico mensageiro (ARNm) da GTPCH e a concentração intracelular de BH439,40. Estes fármacos aumentaram ainda a transcrição da eNOS39,40. Num outro estudo, o tratamento com atorvastatina, pravastatina ou pitavastatina aumentou também a expressão da eNOS, pela estimulação da fosforilação da cínase B de proteínas (Akt) no resíduo Ser473, inibindo a senescência endotelial induzida pelo stresse oxidativo41.
O aumento da expressão da eNOS pode também resultar de uma maior estabilidade do seu ARNm em consequência de poliadenilação. A sinvastatina e a rosuvastatina aumentaram significativamente a poliadenilação e, consequentemente, a estabilidade do ARNm da eNOS em células endoteliais de aorta de bovino42 (Figura 4).
 eNOS. As estatinas estimulam a síntese de NO pela
eNOS. As estatinas estimulam a síntese de NO pela Ação das estatinas na eNOS.
As estatinas estimulam a síntese de NO pela eNOS e ao prevenir o desacoplamento desta enzima, evitam a formação de ROS.
Akt – cínase B de proteínas; AMPK – cínase de proteínas ativada pelo monofosfato de adenosina; ARNm – ácido ribonucleico mensageiro; BH4 – tetra‐hidrobiopterina; eNOS – sintetase endotelial do monóxido de azoto; Cadeia PoliA – cadeia poliadenilada; Cav‐1 – caveolina‐1; GTPCH‐1 – ciclohidrolase I do trifosfato de guanosina; NO – monóxido de azoto; O2 – oxigénio; P – fosfato; PI3K – cínase do 3‐fosfatidilinositol; PKA – cínase A de proteínas; ROS – espécies reativas de oxigénio; Ser633 – serina633da eNOS; Ser1177 – serina1177da eNOS.
As estatinas podem ainda contribuir para o aumento da atividade da eNOS por fosforilação da enzima. A fluvastatina ou pitavastatina promoveram a fosforilação da eNOS nos resíduos Ser1177 e/ou Ser633 através da cínase do 3‐fosfatidilinositol (PI3K)/Akt e da cínase A de proteínas (PKA), respetivamente, aumentando a atividade da eNOS em células endoteliais humanas39,43. A cínase de proteínas ativada pelo monofosfato de adenosina (AMPK), quando estimulada pelas estatinas, pode também fosforilar a eNOS na Ser1177, aumentando a sua atividade44. As estatinas podem ainda melhorar a atividade da eNOS, devido ao seu efeito inibitório na expressão da caveolina‐145, uma molécula que interage com a eNOS nas células endoteliais, influenciando negativamente a sua atividade46. Estes fármacos podem ainda aumentar a atividade e acoplamento da eNOS pela redução da concentração sistémica de dimetilarginina assimétrica, um inibidor da eNOS11.
Efeito das estatinas nas NoxComo referido, as Nox são importantes fontes de ROS e contribuem para o stresse oxidativo. A produção de ROS pelas isoformas Nox2 e Nox1 requer a ativação e translocação de proteínas citoplasmáticas como a Rac, a p47phox ou o seu homólogo Noxo1, e a p67phox ou o seu homólogo Noxa1, que depois interagem com as subunidades membranares Nox1 ou gp91phox (Nox2) e p22phox. Na Nox2 há ainda translocação da subunidade p40phox. A ativação e translocação da proteína Rac está dependente da isoprenilação pelo GGPF, cuja síntese ocorre através da via do mevalonato11. A inibição desta via pelas estatinas bloqueia a ativação da Rac, com consequente redução da atividade das Nox1 e Nox211 (Figura 5).
 Nox2 em células endoteliais. A
Nox2 em células endoteliais. A Ação das estatinas na Nox2 em células endoteliais.
A Nox2 é constituída por vários componentes como as subunidades membranares p22phox e Nox2 (gp91phox), e por subunidades citosólicas como a p47phox, a p67phox, a p40phox e a GTP‐Rac. A ativação da Nox requer a ativação e translocação para a membrana da proteína Rac e da p47phox, p67phox e p40phox. A ativação da Rac está dependente da isoprenilação desta proteína pelo GGPF. A inibição da formação deste último pelas estatinas, previne a ativação da Rac e, consequentemente, da Nox2. Outro mecanismo pelo qual as estatinas inibem a Nox2 parece estar associado a alterações da fosforilação da PKC e da translocação membranar da p47phox e é, em parte, mediado pela adiponectina que inibe a translocação da p47phox.
ADP – difosfato de adenosina; ARNm – ácido ribonucleico mensageiro; FPF – farnesilpirofosfato; GGPF – geranilgeranilpirofosfato; GTP – trifosfato de guanosina; HMG‐CoA – hidroximetilglutaril Coenzima A; Nox – NADPH oxídase; O2 – oxigénio; O2•− – superóxido; PKC – cínase C de proteínas.
O efeito inibitório das estatinas na atividade das Nox parece também envolver efeitos noutras subunidades destas enzimas. Observou‐se a diminuição da expressão do ARNm da p22phox, gp91phox e Nox1, da expressão proteica da p47phox e da translocação da p47phox e p67phox após tratamento com estatinas. Estas inibiram ainda a expressão do recetor AT1 da angiotensina II, que medeia o efeito estimulador da angiotensina II na atividade das Nox11.
Recentemente, foi sugerido que o efeito inibitório das estatinas nas Nox possa ser, em parte, mediado pela adiponectina, uma proteína sintetizada pelos adipócitos. De facto, em doentes com hipercolesterolemia, verificou‐se que o aumento de adiponectina, causado pelo tratamento com atorvastatina, estava associado a redução da forma solúvel da gp91phox (sgp91phox), da produção plaquetar de ROS e de isoprostanos urinários. Observou‐se ainda que o tratamento in vitro com adiponectina impediu a translocação da p47phox e a clivagem da sgp91phox, inibindo a ativação da Nox nas plaquetas47.
Efeito das estatinas nos sistemas antioxidantesAs estatinas podem também estimular defesas antioxidantes. O tratamento in vitro de células endoteliais humanas com atorvastatina induziu o aumento da expressão da catálase e da MnSOD subsequentemente à fosforilação do resíduo Ser473da Akt41. Em células tumorais, a fluvastatina aumentou a expressão do ARNm e da proteína da MnSOD e duplicou a atividade desta enzima48. Estes efeitos na MnSOD parecem dever‐se à diminuição da expressão de um regulador negativo da transcrição desta enzima, a proteína de tipo 2 de ligação a danos no ADN (DNA Damage Binding Protein 2, DDB2)48. A fluvastatina aumentou ainda a atividade da catálase e a concentração de GSH48. O aumento da GSH foi também observado em células promielocíticas humanas tratadas com rosuvastatina e parece resultar do aumento do ARNm e da atividade da sintétase da γ‐glutamilcisteína, a enzima limitante da taxa de síntese da GSH49.
As estatinas afetam ainda outras enzimas antioxidantes, como a tiorredoxina 1 e a HO‐1. Em células endoteliais humanas, o tratamento com atorvastatina estimulou a síntese de NO e consequente S‐nitrosilação da tiorredoxina‐1, aumentando a sua atividade enzimática e promovendo a diminuição das ROS intracelulares27. Por sua vez, o tratamento com rosuvastatina aumentou a expressão do ARNm e da proteína da HO‐150.
Outro efeito protetor das estatinas poderá estar relacionado com a sua ação na PON‐1, uma enzima envolvida no efeito antioxidante da HDL. Em indivíduos com hipercolesterolémia, a atorvastatina aumentou a atividade da PON‐151.
Outros efeitos antioxidantes/anti‐inflamatórios das estatinasA inter‐relação entre stresse oxidativo e inflamação tem sido evidenciada na IC e noutras doenças crónicas, contribuindo para a sua génese e progressão52,53. De facto, o stresse oxidativo ativa vários fatores de transcrição, como, por exemplo, o NF‐κB e a AP‐1, que promovem a expressão de citocinas pró‐inflamatórias, quimiocinas e moléculas de adesão53,54. Por outro lado, a produção de elevadas quantidades de ROS é uma característica das células inflamatórias ativadas53. Algumas citocinas promovem também a síntese de ROS em células endoteliais e musculares lisas vasculares55,56.
As estatinas exercem efeitos em várias moléculas que desempenham, simultaneamente, um papel fundamental no stresse oxidativo e inflamação. Por exemplo, a MPO, presente nos neutrófilos e monócitos, contribui para a formação de ROS durante os processos inflamatórios, promovendo a oxidação lipídica e lesão tecidual57. A MPO interfere com a homeostasia endotelial, contribuindo para a iniciação e progressão da aterosclerose57 e está também associada a um agravamento da classe funcional New York Heart Association (NYHA) na IC16. Há evidência de que as estatinas inibem, significativamente, a expressão do ARNm da MPO em monócitos‐macrófagos humanos ou de murganho. Este efeito está dependente do bloqueio da via do mevalonato e redução da formação de GGPF58.
A galectina‐3, uma proteína da família das lectinas, é também um biomarcador associado ao stresse oxidativo e à inflamação. Esta molécula está envolvida na proliferação, quimiotaxia, fagocitose, apoptose, angiogénese e fibrose miocárdica, mecanismos que participam na patogénese das doenças cardiovasculares (DCV). A libertação de galectina‐3 pelos monócitos e macrófagos pode ser regulada pelas ROS e pelas Nox. Curiosamente, in vitro, a galectina‐3 estimula a síntese de O2•− pelos monócitos e macrófagos, logo é possível que contribua para o ciclo vicioso inerente ao stresse oxidativo e inflamação59. Num modelo experimental de aterosclerose, verificou‐se que a expressão de galectina‐3 aumentou proporcionalmente à extensão e grau de inflamação da placa de ateroma, e que a atorvastatina reduziu marcadamente a expressão de galectina‐3 e a quantidade de macrófagos da placa60.
Recentemente, demonstrou‐se que as estatinas podem promover a resolução da inflamação pela estimulação da síntese de 15‐epi‐lipoxina A4 (15‐epi‐LXA4), que possui propriedades pró‐resolutivas, anti‐inflamatórias, antioxidantes, vasodilatadoras e antiproliferativas 61–66 (Figura 6). A síntese de 15‐epi‐LXA4 envolve a ação sequencial da cicloxigenase‐2 (COX‐2) e da 5‐lipoxigenase (5‐LOX), a partir do ácido araquidónico. As estatinas estimulam a expressão e a S‐nitrosilação da COX‐2 e, consequentemente, a síntese do ácido 15R‐hidroxieicosatetraenóico que é depois convertido em 15‐epi‐LXA4 pela 5‐LOX (Figura 6)61. O tratamento de ratos com atorvastatina aumentou significativamente a síntese de 15‐epi‐LXA4 no miocárdio, através da S‐nitrosilação da COX‐262. Num modelo animal de inflamação das vias respiratórias, o tratamento com lovastatina promoveu também a formação desta lipoxina e reduziu marcadamente a inflamação pulmonar aguda. Adicionalmente, o tratamento in vitro com lovastatina aumentou a produção de 15‐epi‐LXA4, durante a interação celular entre células polimorfonucleares e células epiteliais das vias respiratórias humanas estimuladas com citocinas63 (Figura 7).
 MMP3 – metaloproteinase da matriz 3; NO – monóxido de azoto; O2•− – superóxido; ONOO− – peroxinitrito; PGI2 – prostaciclina;
MMP3 – metaloproteinase da matriz 3; NO – monóxido de azoto; O2•− – superóxido; ONOO− – peroxinitrito; PGI2 – prostaciclina; Estatinas e síntese de 15‐epi‐lipoxinas: efeitos protetores.
A S‐nitrosilação da COX‐2 pelas estatinas promove a síntese de 15‐epi‐lipoxinas, que exercem efeitos protetores em diferentes tipos de células, contribuindo para a resolução da inflamação.
AA – ácido araquidónico; CCR5 – recetor de tipo 5 de quimiocinas C‐C; CD11B/CD18 – recetor do complemento 3 (cluster of differentiation molecule 11B/Integrin beta‐2); COX‐2 – ciclooxigenase‐2; HO‐1 – heme oxigenase‐1; ICAM‐1 – molécula de adesão intercelular ‐1; MMP3 – metaloproteinase da matriz 3; NO – monóxido de azoto; O2•− – superóxido; ONOO− – peroxinitrito; PGI2 – prostaciclina; ROS – espécies reativas de oxigénio; TNF‐α – fator de necrose tumoral alfa; VEFG – fator de crescimento endotelial vascular; 5‐LOX – 5‐lipoxigenase; 15R‐HETE – ácido 15R‐hidroxieicosatetraenóico; 15‐epi‐LXA4 – 15‐epi‐lipoxina A4.
 ARNm – ácido ribonucleico mensageiro;
ARNm – ácido ribonucleico mensageiro; Resumo de efeitos antioxidantes e anti‐inflamatórios das estatinas.
ADMA – dimetilarginina assimétrica; ARNm – ácido ribonucleico mensageiro; BH4 – tetra‐hidrobiopterina; eNOS – sintetase endotelial de monóxido de azoto; GSH – glutationa; GTPCH – ciclohidrolase I do trifosfato de guanosina; HDL – lipoproteína de elevada densidade; HO‐1 – heme oxigenase‐1; MnSOD – superóxido dismútase mitocondrial; MPO – mieloperoxídase; Nox – NADPH oxídase; PON‐1 – Paraoxonase‐1; 15‐epi‐LXA4 – 15‐epi‐lipoxina A4.
O stresse oxidativo está associado ao envelhecimento e ao desenvolvimento de múltiplas patologias, entre as quais as DCV14. Na última década, vários estudos avaliaram os efeitos das estatinas em marcadores do stresse oxidativo e da função endotelial em patologias cardiovasculares, renais e metabólicas.
Em doentes com IC com fração de ejeção (FE) ventricular reduzida, o tratamento durante um mês com sinvastatina ou atorvastatina reduziu a produção de ROS e a concentração sistémica de malonildialdeído (MDA), um marcador de peroxidação lipídica, aumentou a atividade da EC‐SOD e melhorou a função endotelial e a capacidade funcional67,68. Noutro estudo em doentes com IC sistólica, o tratamento durante um mês com rosuvastatina reduziu também significativamente a concentração plasmática de MPO e de oxLDL69.
Em indivíduos com DC submetidos a cirurgia de revascularização coronária, o tratamento prévio com atorvastatina ou pravastatina, durante quatro semanas, reduziu de forma significativa a expressão do ARNm e a atividade da Rac1 em amostras de tecido miocárdico. Adicionalmente, estes tratamentos diminuíram a atividade miocárdica das Nox induzida pela angiotensina II70. Noutro estudo, efetuado também em doentes submetidos a cirurgia cardíaca e monitorizados até à alta hospitalar, verificou‐se uma forte associação entre a produção de O2•− e peroxinitrito e as complicações pós‐operatórias durante o período de internamento. O tratamento pré‐operatório, durante três dias, com atorvastatina diminuiu significativamente a atividade das Nox e a produção de O2•− e peroxinitrito no tecido miocárdico. Adicionalmente, a incubação ex vivo de tecido miocárdico com atorvastatina causou uma diminuição da atividade da Rac1 e das Nox, que foi revertida pela adição de mevalonato71. Outro estudo semelhante demonstrou que a administração pré‐operatória de atorvastatina aumentou a disponibilidade de BH4 e reduziu quer a produção basal de O2•−, quer a atribuível à eNOS desacoplada, em amostras de artérias mamárias internas72. A terapêutica com estatinas parece ainda diminuir a concentração plasmática de MPO em doentes com síndrome coronária aguda, mas não em indivíduos com DC estável73.
Os efeitos redox das estatinas têm sido também avaliados em indivíduos com hiperlipidemia, tendo sido observados vários efeitos protetores, tais como a redução da produção de ROS e aumento da síntese de NO pelas plaquetas, a redução de isoprostanos urinários e plaquetares, a redução da ativação da Rac1 plaquetar, a inibição da Nox2 sérica e plaquetar, o aumento de adiponectina e consequente redução da gp91phox e da atividade da Nox, o aumento da concentração plasmática de vitamina E e a redução de danos do ADN em indivíduos hipercolesterolémicos com polimorfismo C242T do gene da p22phox, associado ao risco de desenvolvimento de DC47,74–78.
Uma vez que o stresse oxidativo está também associado à patogénese da fibrilhação auricular (FA), um estudo recente avaliou os efeitos das estatinas na produção de ROS quer em doentes que desenvolveram FA após cirurgia cardíaca quer em doentes com FA recorrente. A atorvastatina reduziu a ativação da Rac1 e Nox em aurículas direitas de doentes com FA pós‐operatória, mas não alterou a produção de ROS, o desacoplamento da eNOS ou a quantidade de BH4 em doentes com FA recorrente. Estes resultados sugerem que a indução da Nox é um evento precoce, mas transitório na fisiopatologia desta arritmia79.
As estatinas parecem também exercer um efeito protetor na diabetes mellitus. Em doentes com nefropatia diabética, o tratamento com rosuvastatina durante seis meses melhorou a função renal e diminuiu significativamente a concentração sérica de produtos de peroxidação lipídica e a excreção urinária de um marcador de oxidação do ADN, a 8‐hidroxi‐2′‐desoxiguanosina (8‐OHdG)80. Na polineuropatia diabética, o tratamento com rosuvastatina, durante 12 semanas, causou uma melhoria na gravidade, sintomas e parâmetros de condução nervosa, bem como uma redução da peroxidação lipídica81.
Apesar da crescente evidência dos efeitos antioxidantes das estatinas em patologias cardiovasculares e metabólicas, há também alguns estudos em que esses efeitos não se observaram. Em indivíduos com elevado risco de DCV, os tratamentos com atorvastatina ou sinvastatina não alteraram significativamente a concentração plasmática de MDA e MPO, nem a excreção urinária de 8‐OHdG82 e, noutro estudo em doentes com nefropatia diabética, o tratamento com atorvastatina não melhorou a disponibilidade de NO83.
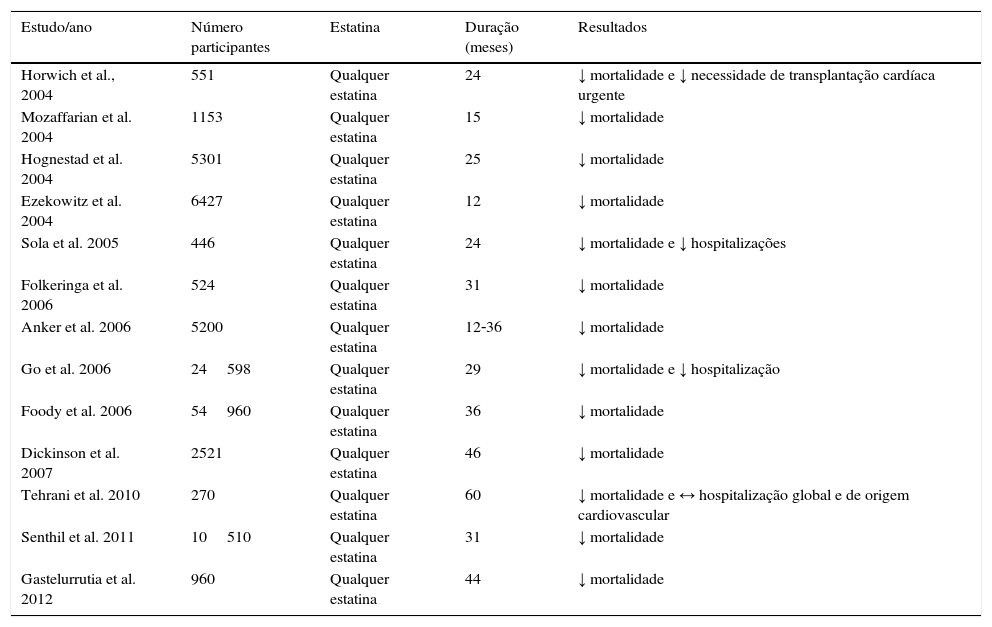
Estatinas na insuficiência cardíaca crónica: ensaios clínicosA maioria dos estudos referentes aos efeitos das estatinas na IC são observacionais, não aleatorizados e referentes à IC com FE reduzida (FE ≤35%). Estes sugerem que as estatinas exercem um efeito benéfico na mortalidade (Tabela 2). Na IC com FE preservada verificou‐se também uma diminuição da mortalidade após tratamento com estatinas84.
Efeito das estatinas na IC: estudos observacionais não randomizados. Adaptado de6–8,84,90
| Estudo/ano | Número participantes | Estatina | Duração (meses) | Resultados |
|---|---|---|---|---|
| Horwich et al., 2004 | 551 | Qualquer estatina | 24 | ↓ mortalidade e ↓ necessidade de transplantação cardíaca urgente |
| Mozaffarian et al. 2004 | 1153 | Qualquer estatina | 15 | ↓ mortalidade |
| Hognestad et al. 2004 | 5301 | Qualquer estatina | 25 | ↓ mortalidade |
| Ezekowitz et al. 2004 | 6427 | Qualquer estatina | 12 | ↓ mortalidade |
| Sola et al. 2005 | 446 | Qualquer estatina | 24 | ↓ mortalidade e ↓ hospitalizações |
| Folkeringa et al. 2006 | 524 | Qualquer estatina | 31 | ↓ mortalidade |
| Anker et al. 2006 | 5200 | Qualquer estatina | 12‐36 | ↓ mortalidade |
| Go et al. 2006 | 24598 | Qualquer estatina | 29 | ↓ mortalidade e ↓ hospitalização |
| Foody et al. 2006 | 54960 | Qualquer estatina | 36 | ↓ mortalidade |
| Dickinson et al. 2007 | 2521 | Qualquer estatina | 46 | ↓ mortalidade |
| Tehrani et al. 2010 | 270 | Qualquer estatina | 60 | ↓ mortalidade e ↔ hospitalização global e de origem cardiovascular |
| Senthil et al. 2011 | 10510 | Qualquer estatina | 31 | ↓ mortalidade |
| Gastelurrutia et al. 2012 | 960 | Qualquer estatina | 44 | ↓ mortalidade |
↓ – diminuição; ↑ – aumento; ↔ – manutenção.
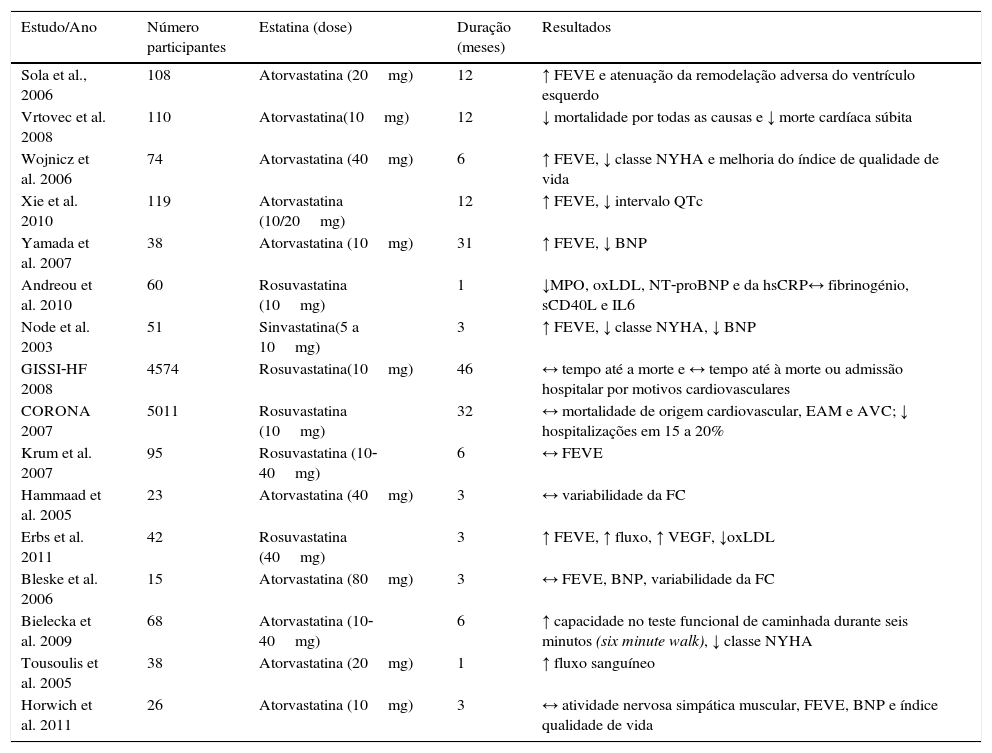
Entretanto surgiram vários ensaios clínicos aleatorizados (Tabela 3), sendo o CORONA (Controlled Rosuvastatin Multinational Trial in Heart Failure) e o GISSI‐HF (Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto miocardico – Heart Failure) aqueles com maior número de doentes85,86.
Efeito das estatinas na IC com fração de ejeção reduzida: estudos aleatorizados. Adaptado de6‐8,69,85,87
| Estudo/Ano | Número participantes | Estatina (dose) | Duração (meses) | Resultados |
|---|---|---|---|---|
| Sola et al., 2006 | 108 | Atorvastatina (20mg) | 12 | ↑ FEVE e atenuação da remodelação adversa do ventrículo esquerdo |
| Vrtovec et al. 2008 | 110 | Atorvastatina(10mg) | 12 | ↓ mortalidade por todas as causas e ↓ morte cardíaca súbita |
| Wojnicz et al. 2006 | 74 | Atorvastatina (40mg) | 6 | ↑ FEVE, ↓ classe NYHA e melhoria do índice de qualidade de vida |
| Xie et al. 2010 | 119 | Atorvastatina (10/20mg) | 12 | ↑ FEVE, ↓ intervalo QTc |
| Yamada et al. 2007 | 38 | Atorvastatina (10mg) | 31 | ↑ FEVE, ↓ BNP |
| Andreou et al. 2010 | 60 | Rosuvastatina (10mg) | 1 | ↓MPO, oxLDL, NT‐proBNP e da hsCRP↔ fibrinogénio, sCD40L e IL6 |
| Node et al. 2003 | 51 | Sinvastatina(5 a 10mg) | 3 | ↑ FEVE, ↓ classe NYHA, ↓ BNP |
| GISSI‐HF 2008 | 4574 | Rosuvastatina(10mg) | 46 | ↔ tempo até a morte e ↔ tempo até à morte ou admissão hospitalar por motivos cardiovasculares |
| CORONA 2007 | 5011 | Rosuvastatina (10mg) | 32 | ↔ mortalidade de origem cardiovascular, EAM e AVC; ↓ hospitalizações em 15 a 20% |
| Krum et al. 2007 | 95 | Rosuvastatina (10‐40mg) | 6 | ↔ FEVE |
| Hammaad et al. 2005 | 23 | Atorvastatina (40mg) | 3 | ↔ variabilidade da FC |
| Erbs et al. 2011 | 42 | Rosuvastatina (40mg) | 3 | ↑ FEVE, ↑ fluxo, ↑ VEGF, ↓oxLDL |
| Bleske et al. 2006 | 15 | Atorvastatina (80mg) | 3 | ↔ FEVE, BNP, variabilidade da FC |
| Bielecka et al. 2009 | 68 | Atorvastatina (10‐40mg) | 6 | ↑ capacidade no teste funcional de caminhada durante seis minutos (six minute walk), ↓ classe NYHA |
| Tousoulis et al. 2005 | 38 | Atorvastatina (20mg) | 1 | ↑ fluxo sanguíneo |
| Horwich et al. 2011 | 26 | Atorvastatina (10mg) | 3 | ↔ atividade nervosa simpática muscular, FEVE, BNP e índice qualidade de vida |
↓ – diminuição; ↑ – aumento; ↔ – manutenção; AVC – acidente vascular cerebral; BNP – peptídeo natriurético de tipo B; EAM – enfarte agudo do miocárdio; FC – frequência cardíaca; FEVE – fração de ejeção ventricular esquerda; hsCRP – proteína C reativa de alta sensibilidade; IL6 – Interleucina 6; MPO – mieloperoxídase; NYHA – New York Heart Association; NT‐proBNP – N‐terminal do pro‐peptídeo natriurético de tipo B; oxLDL – lipoproteínas de baixa densidade oxidadas; QTc – intervalo QT corrigido; sCD40L – forma solúvel do CD40 ligando; VEGF – fator de crescimento vascular endotelial.
O estudo CORONA é um ensaio clínico prospetivo e aleatorizado, no qual participaram 5011 doentes com IC sintomática (NYHA II‐IV), de etiologia isquémica, com FE ventricular esquerda (FEVE) ≤0,40 (NYHA III ou IV) ou ≤0,35 (NYHA II) e idade ≥60 anos86. Apesar dos benefícios no perfil lipídico e da redução da proteína C reativa de alta sensibilidade, não se observou uma redução na mortalidade de origem cardiovascular, no enfarte agudo do miocárdio (EAM) e no acidente vascular cerebral (AVC)87.
O GISSI‐HF é um estudo prospetivo e aleatorizado no qual participaram 4574 doentes com IC (NYHA II‐IV), independentemente da etiologia ou FEVE, com idade ≥18 anos. Verificou‐se que, apesar da administração diária da rosuvastatina poder ser feita com segurança, esta não estava associada à redução dos eventos clínicos (tempo até à morte e o tempo até à admissão hospitalar por motivos cardiovasculares) durante o período de seguimento de 46 meses85.
Nesse contexto, foram propostas várias explicações para os resultados obtidos no CORONA e no GISSI‐HF. É possível que os efeitos das estatinas não sejam dependentes da classe terapêutica porque a atorvastatina, por oposição à rosuvastatina, diminuiu a mortalidade global, a admissão hospitalar, melhorou a FEVE88 e reduziu a concentração sistémica de peptídeo natriurético de tipo B (BNP), o que poderá ser explicado pelas diferentes propriedades farmacocinéticas das estatinas89. Além disso, uma meta‐análise não identificou nenhuma correlação entre a dose e os resultados obtidos, sugerindo que o tipo de estatina talvez seja mais importante que a dose88.
Possivelmente, as características da população estudada, nomeadamente a idade (no CORONA e no GISSI‐HF, a média da idade foi de 73 e 68 anos, respetivamente) e a exclusão de doentes medicados previamente com estatinas também interferiram com os resultados90.
Segundo uma teoria alternativa, as estatinas poderão ser benéficas nos estadios iniciais da IC, mas, nas fases mais avançadas, não impedirão a deterioração progressiva da função cardíaca91. Assim, poderá ser útil identificar previamente os subgrupos de doentes que obterão maior benefício com estes fármacos. Doentes medicados com rosuvastatina e valores do segmento N‐terminal da pró‐hormona do BNP (NT‐proBNP) no menor tercil (<103pmol/l ou <868pg/ml), parecem apresentar uma maior redução da mortalidade cardiovascular, EAM e AVC92. Da mesma forma, doentes medicados com rosuvastatina e concentrações de galectina‐3 ≤19ng/mL apresentaram uma taxa de eventos primários, mortalidade total ou de qualquer causa e hospitalizações por IC inferiores comparativamente ao placebo. Doentes com concentrações superiores de galectina‐3 parecem não obter semelhantes benefícios93.
Atualmente, a Sociedade Europeia de Cardiologia (ESC) e o Colégio Americano de Cardiologia (ACC) não recomendam a prescrição de estatinas como terapêutica adjuvante da IC na ausência de outras indicações para o seu uso2,3.
ConclusãoPara além dos seus efeitos na síntese de colesterol, as estatinas possuem propriedades pleiotrópicas, entre as quais são de destacar as ações antioxidantes. Estes efeitos parecem resultar do bloqueio da síntese de intermediários isoprenoides na via do mevalonato.
Atualmente é reconhecida a importância do stresse oxidativo na patogénese de muitas doenças, incluindo a IC. As ROS interferem em vários processos que afetam a estrutura e função cardíacas, contribuindo para a apoptose e disfunção mitocondrial dos cardiomiócitos, disfunção contráctil, fibrose e hipertrofia do miocárdio e ainda para a disfunção endotelial e patogénese da aterosclerose. As estatinas podem diminuir a concentração de ROS e aumentar a síntese de NO, promovendo deste modo o equilíbrio redox cardíaco. Estes efeitos são mediados principalmente pela inibição da ativação de enzimas pró‐oxidantes como as Nox e pela estimulação da expressão, atividade e estabilidade da eNOS. Adicionalmente, as estatinas podem estimular outras enzimas antioxidantes e contribuir para o controlo de processos inflamatórios. De facto, estes fármacos exercem efeitos em várias moléculas que desempenham, simultaneamente, um papel fundamental no stresse oxidativo e inflamação, salientando‐se a estimulação da síntese da 15‐epi‐LXA4, um mediador com propriedades pró‐resolutivas, anti‐inflamatórias e antioxidantes. Os efeitos antioxidantes das estatinas têm sido demonstrados em várias patologias cardiovasculares no Homem, incluindo a IC, a DC e a FA, principalmente nas fases iniciais destas doenças. Relativamente aos efeitos destes fármacos na redução de eventos cardiovasculares e mortalidade na IC, apesar de vários estudos observacionais demonstrarem uma diminuição da mortalidade após terapêutica com estatinas, dois grandes ensaios clínicos, o CORONA e o GISSI‐HF, não constataram o mesmo benefício. Atualmente, a ESC e o ACC não recomendam o tratamento com estatinas na IC, na ausência de outras indicações para a sua prescrição. Face à inexistência de resultados consensuais, sugere‐se a realização de novos ensaios clínicos para esclarecer a relevância clínica das estatinas em subgrupos de doentes com IC e a sua relação com os efeitos no estado redox.
Responsabilidades éticasProteção de pessoas e animaisOs autores declaram que para esta investigação não se realizaram experiências em seres humanos e/ou animais.
Confidencialidade dos dadosOs autores declaram que não aparecem dados de pacientes neste artigo.
Direito à privacidade e consentimento escritoOs autores declaram que não aparecem dados de pacientes neste artigo.
Conflito de interessesOs autores declaram não haver conflito de interesses.
