







Statins are the most commonly prescribed drugs for the treatment of dyslipidemia. They are also recommended in primary and secondary prevention of cardiovascular disease. In addition to decreasing cholesterol synthesis, statins interfere with the synthesis of isoprenoid intermediates, which may explain many of their pleiotropic properties, including their antioxidant effects.
Oxidative stress is defined as an imbalance between the synthesis of reactive oxygen species and their elimination by antioxidant defense systems, with a prevailing pro-oxidant status that results in macromolecular damage and disruption of cellular redox signaling. Reactive oxygen species interfere with various processes that affect cardiac structure and function, contributing to the contractile dysfunction, myocardial hypertrophy and fibrosis observed in the pathophysiology of heart failure. By regulating several molecular pathways that control nicotinamide adenine dinucleotide phosphate oxidase and endothelial nitric oxide synthase activity, statins help restore redox homeostasis. These drugs also contribute to the control of inflammation and appear to have a protective role in various diseases. The results of observational studies and clinical trials with statins in heart failure have not been consensual.
This review aims to analyze the role of oxidative stress in heart failure and the molecular mechanisms underlying statins’ antioxidant properties. It also examines current scientific evidence on the use of these drugs as a specific treatment for heart failure.
As estatinas são os fármacos mais prescritos no tratamento da dislipidemia, estando recomendadas na prevenção primária e secundária das doenças cardiovasculares. Para além de diminuírem a síntese de colesterol, interferem com a síntese de intermediários isoprenoides, o que poderá explicar muitos dos seus efeitos pleiotrópicos, incluindo a ação antioxidante.
O stresse oxidativo é definido como um desequilíbrio entre a formação de espécies reativas de oxigénio e a sua eliminação por sistemas de defesa antioxidantes, com prevalência de um estado pró-oxidante com efeitos deletérios nas macromoléculas orgânicas e na sinalização redox celular. As espécies reativas de oxigénio podem interferir com vários processos que afetam a estrutura e função cardíacas, contribuindo para a disfunção contráctil, fibrose e hipertrofia do miocárdio observadas na fisiopatologia da insuficiência cardíaca. As estatinas promovem a restauração do equilíbrio redox pela regulação de várias vias moleculares responsáveis pelo controlo da atividade de enzimas como a NADPH oxídase e a sintetase endotelial de monóxido de azoto. Estes fármacos contribuem também para o controlo de processos inflamatórios e parecem desempenhar um papel protetor em várias patologias. Os resultados de estudos observacionais e ensaios clínicos, realizados com o objetivo de esclarecer o efeito das estatinas na insuficiência cardíaca, não têm sido consensuais. Esta revisão tem como principal objetivo analisar o papel do stresse oxidativo na insuficiência cardíaca e os mecanismos moleculares inerentes às propriedades antioxidantes das estatinas. Pretende ainda reunir a evidência científica atual relativa à utilização destes fármacos como terapêutica específica da insuficiência cardíaca.
8-hydroxy-2′-deoxyguanosine
15-epi-lipoxin A4
5-lipoxygenase
American College of Cardiology
atrial fibrillation
protein kinase B
adenosine monophosphate-activated protein kinase
activator protein 1
apoptosis signal-regulating kinase 1
tetrahydrobiopterin
brain-type natriuretic peptide
coronary artery disease
Controlled Rosuvastatin Multinational Trial in Heart Failure
cyclooxygenase 2
cardiovascular disease
Copper, zinc superoxide dismutase (cytoplasmic isoform; SOD-1)
DNA damage-binding protein 2
extracellular superoxide dismutase (SOD-3)
endothelial nitric oxide synthase
extracellular signal-regulated kinases 1 and 2
European Society of Cardiology
farnesyl pyrophosphate
geranylgeranyl pyrophosphate
Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto miocardico-Heart Failure
glutathione peroxidase
glutathione
guanosine triphosphate
guanosine triphosphate cyclohydrolase
hydrogen peroxide
hypochlorous acid
high-density lipoprotein
heart failure
hydroxymethylglutaryl-coenzyme A
heme oxygenase 1
low-density lipoprotein
left ventricular ejection fraction
monoamine oxidase
mitogen-activated protein kinase
malondialdehyde
myocardial infarction
matrix metalloproteinases
manganese-dependent superoxide dismutase (mitochondrial isoform; SOD-2)
myeloperoxidase
messenger ribonucleic acid
nicotinamide adenine dinucleotide phosphate
nuclear factor kappa B
nitric oxide
nicotinamide adenine dinucleotide phosphate oxidases
N-terminal brain-type natriuretic peptide
New York Heart Association
superoxide
oxidized LDL
phosphoinositide 3-kinase
protein kinase A
reactive oxygen species
serine 1177
serine 473
serine 633
sarcoplasmic Ca2+-ATPase
superoxide dismutase
xanthine oxidase
The prevalence of heart failure (HF) in the USA is predicted to increase by 46% between 2012 and 2030.1 In Portugal, mortality rates remain at approximately 50% within five years of diagnosis.2 It is therefore essential to explore the main pathophysiological pathways of HF and to review current therapies.
HF is a complex clinical syndrome that can be defined as an abnormality of cardiac structure or function affecting ventricular filling and ejection,3 in which there is evidence of imbalances in redox status and nitric oxide (NO) production that can be redressed by appropriate therapy.4
In Portugal, hydroxymethylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors or statins (Figure 1) account for 90% of lipid-lowering drugs prescribed,5 but it is not known what proportion are prescribed in HF. Although various observational studies suggest that statins may reduce the morbidity and mortality associated with HF, due in part to their antioxidant properties,6–8 and that coronary artery disease (CAD), in which the value of statin therapy is well established, is responsible for around two-thirds of cases of systolic HF,3 the benefit of statins specifically for HF has been called into question by the results of randomized clinical trials. Their use is also limited by cardiac cachexia and by the paradoxical association of low serum cholesterol with worse HF prognosis.9
 HMG-CoA reductase inhibitors (statins) (adapted from94). Lovastatin, simvastatin and pravastatin are of fungal origin, while more recent statins are completely synthetic. Statins of fungal origin are structurally related and have a hydronaphthalene ring in common. Lovastatin and simvastatin are administered as inactive prodrugs while pravastatin is given in the active form. Totally synthetic statins have different structures that may account for their solubility differences in water.' title='Chemical structure of
HMG-CoA reductase inhibitors (statins) (adapted from94). Lovastatin, simvastatin and pravastatin are of fungal origin, while more recent statins are completely synthetic. Statins of fungal origin are structurally related and have a hydronaphthalene ring in common. Lovastatin and simvastatin are administered as inactive prodrugs while pravastatin is given in the active form. Totally synthetic statins have different structures that may account for their solubility differences in water.' title='Chemical structure of Chemical structure of HMG-CoA reductase inhibitors (statins) (adapted from94).
Lovastatin, simvastatin and pravastatin are of fungal origin, while more recent statins are completely synthetic. Statins of fungal origin are structurally related and have a hydronaphthalene ring in common. Lovastatin and simvastatin are administered as inactive prodrugs while pravastatin is given in the active form. Totally synthetic statins have different structures that may account for their solubility differences in water.
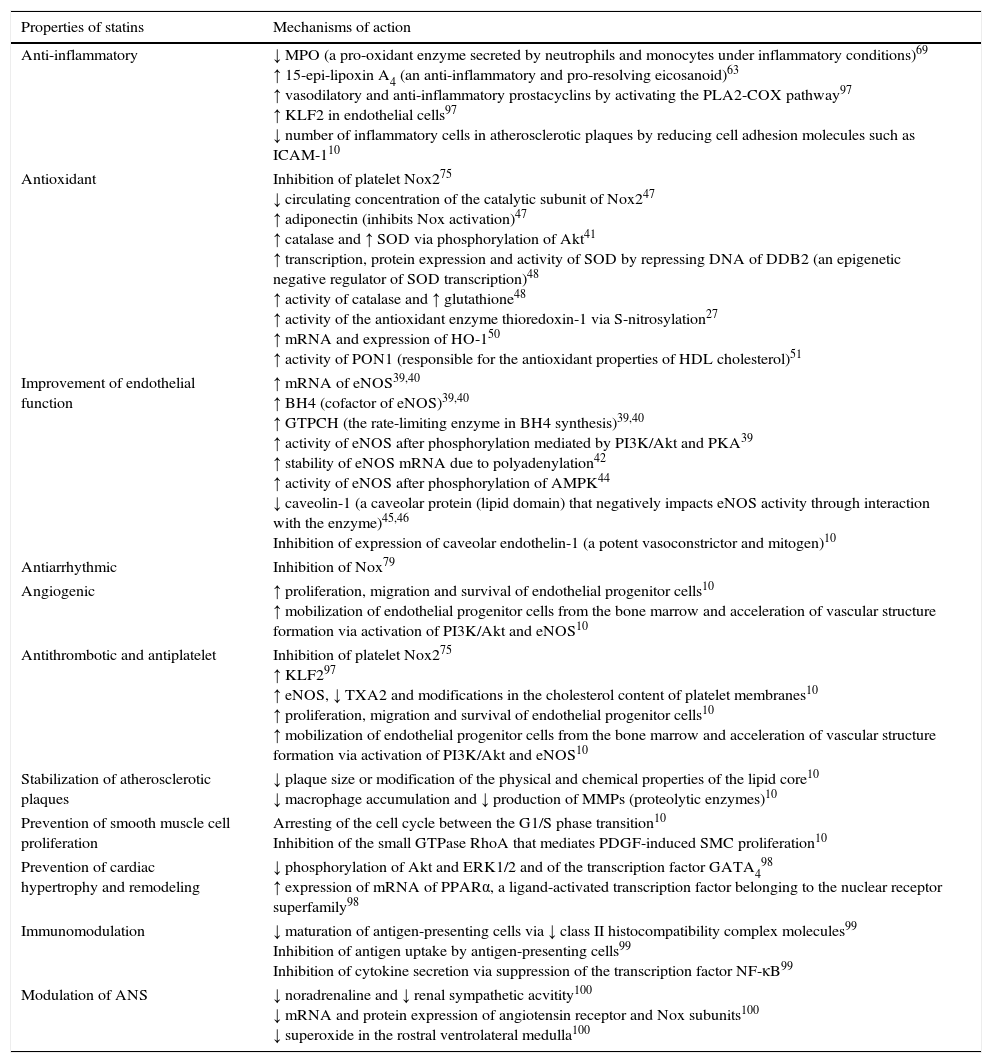
Statins’ main mechanism of action is inhibition of HMG-CoA reductase, the rate-limiting enzyme of cholesterol synthesis. Inhibition of this enzyme interferes with the synthesis of mevalonate and isoprenoid intermediates, particularly farnesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP), which are responsible for the isoprenylation of a wide range of proteins including small proteins associated with guanosine triphosphate (GTP) such as Ras, Rho and Rac (Figure 2). Isoprenylation of these molecules is essential for the covalent attachment, subcellular localization, and intracellular trafficking of membrane-associated proteins.10 Inhibition of the synthesis of isoprenoid intermediates may underlie many of statins’ pleiotropic effects (Table 1),11 including their antioxidant and anti-inflammatory action.
 eNOS: endothelial nitric oxide synthase; ET-1: endothelin-1;
eNOS: endothelial nitric oxide synthase; ET-1: endothelin-1; Effects of statins on the mevalonate pathway (adapted from95,96). ↑: increased; ↓: decreased; eNOS: endothelial nitric oxide synthase; ET-1: endothelin-1; HMG-CoA: hydroxymethylglutaryl-coenzyme A; PAI-1: plasminogen activator inhibitor-1; PP: pyrophosphate; t-PA: tissue plasminogen activator.
Pleiotropic effects of statins and associated molecular pathways.
| Properties of statins | Mechanisms of action |
|---|---|
| Anti-inflammatory | ↓ MPO (a pro-oxidant enzyme secreted by neutrophils and monocytes under inflammatory conditions)69 ↑ 15-epi-lipoxin A4 (an anti-inflammatory and pro-resolving eicosanoid)63 ↑ vasodilatory and anti-inflammatory prostacyclins by activating the PLA2-COX pathway97 ↑ KLF2 in endothelial cells97 ↓ number of inflammatory cells in atherosclerotic plaques by reducing cell adhesion molecules such as ICAM-110 |
| Antioxidant | Inhibition of platelet Nox275 ↓ circulating concentration of the catalytic subunit of Nox247 ↑ adiponectin (inhibits Nox activation)47 ↑ catalase and ↑ SOD via phosphorylation of Akt41 ↑ transcription, protein expression and activity of SOD by repressing DNA of DDB2 (an epigenetic negative regulator of SOD transcription)48 ↑ activity of catalase and ↑ glutathione48 ↑ activity of the antioxidant enzyme thioredoxin-1 via S-nitrosylation27 ↑ mRNA and expression of HO-150 ↑ activity of PON1 (responsible for the antioxidant properties of HDL cholesterol)51 |
| Improvement of endothelial function | ↑ mRNA of eNOS39,40 ↑ BH4 (cofactor of eNOS)39,40 ↑ GTPCH (the rate-limiting enzyme in BH4 synthesis)39,40 ↑ activity of eNOS after phosphorylation mediated by PI3K/Akt and PKA39 ↑ stability of eNOS mRNA due to polyadenylation42 ↑ activity of eNOS after phosphorylation of AMPK44 ↓ caveolin-1 (a caveolar protein (lipid domain) that negatively impacts eNOS activity through interaction with the enzyme)45,46 Inhibition of expression of caveolar endothelin-1 (a potent vasoconstrictor and mitogen)10 |
| Antiarrhythmic | Inhibition of Nox79 |
| Angiogenic | ↑ proliferation, migration and survival of endothelial progenitor cells10 ↑ mobilization of endothelial progenitor cells from the bone marrow and acceleration of vascular structure formation via activation of PI3K/Akt and eNOS10 |
| Antithrombotic and antiplatelet | Inhibition of platelet Nox275 ↑ KLF297 ↑ eNOS, ↓ TXA2 and modifications in the cholesterol content of platelet membranes10 ↑ proliferation, migration and survival of endothelial progenitor cells10 ↑ mobilization of endothelial progenitor cells from the bone marrow and acceleration of vascular structure formation via activation of PI3K/Akt and eNOS10 |
| Stabilization of atherosclerotic plaques | ↓ plaque size or modification of the physical and chemical properties of the lipid core10 ↓ macrophage accumulation and ↓ production of MMPs (proteolytic enzymes)10 |
| Prevention of smooth muscle cell proliferation | Arresting of the cell cycle between the G1/S phase transition10 Inhibition of the small GTPase RhoA that mediates PDGF-induced SMC proliferation10 |
| Prevention of cardiac hypertrophy and remodeling | ↓ phosphorylation of Akt and ERK1/2 and of the transcription factor GATA498 ↑ expression of mRNA of PPARα, a ligand-activated transcription factor belonging to the nuclear receptor superfamily98 |
| Immunomodulation | ↓ maturation of antigen-presenting cells via ↓ class II histocompatibility complex molecules99 Inhibition of antigen uptake by antigen-presenting cells99 Inhibition of cytokine secretion via suppression of the transcription factor NF-κB99 |
| Modulation of ANS | ↓ noradrenaline and ↓ renal sympathetic acvitity100 ↓ mRNA and protein expression of angiotensin receptor and Nox subunits100 ↓ superoxide in the rostral ventrolateral medulla100 |
↑: increased; ↓: decreased; Akt: protein kinase B; AMPK: adenosine monophosphate-activated protein kinase; ANS: autonomic nervous system; BH4: tetrahydrobiopterin; COX: cyclooxygenase; DDB2: damage-binding protein 2; eNOS: endothelial nitric oxide synthase; ERK1/2: extracellular signal-regulated kinases 1 and 2; GATA4: transcription factor GATA-4; GTPCH: guanosine triphosphate cyclohydrolase; HDL: high-density lipoprotein; HO-1: heme oxygenase 1; ICAM-1: intercellular adhesion molecule 1; KLF2: Kruppel-like factor 2; MMPs: matrix metalloproteases; MPO: myeloperoxidase; mRNA: messenger RNA; NF-κB: nuclear factor kappa B; NO: nitric oxide; Nox: nicotinamide adenine dinucleotide phosphate oxidases; PDGF: platelet-derived growth factor; PI3K: phosphoinositide 3-kinase; PKA: protein kinase A; PLA2: phospholipase A2; PON1: paraoxonase 1; PPARα: peroxisome proliferator-activated receptor alpha; RhoA: Ras homolog gene family, member A; SMC: smooth muscle cells; SOD: superoxide dismutase; TXA2: thromboxane A2.
The main aim of this review is to analyze the role of oxidative stress in HF and the molecular mechanisms underlying statins’ antioxidant properties. It also examines current scientific evidence on the use of these drugs as a specific treatment for HF.
Heart failure and oxidative stressSeveral experimental and clinical studies in recent years have demonstrated the importance of oxidative stress in the pathogenesis of HF. Oxidative stress is defined as an imbalance between the synthesis of reactive oxygen species (ROS) and their elimination by antioxidant defense systems, with a prevailing pro-oxidant status that results in macromolecular damage and disrupts cellular redox signaling.12 The ROS family includes free radicals such as superoxide (O2−) and the hydroxyl radical (HO) as well as non-radical oxidants such as hydrogen peroxide (H2O2) and hypochlorous acid (HClO).13
Cardiomyocytes, endothelial cells and neutrophils all produce ROS in the heart.14 The principal enzymes involved in this process are mitochondrial oxidases, nicotinamide adenine dinucleotide phosphate oxidases (Nox), xanthine oxidase (XO), endothelial nitric oxide synthase (eNOS), monoamine oxidase (MAO) and myeloperoxidase (MPO).14–16
Mitochondria are the main source of ROS. More than 90% of the total oxygen (O2) consumed by aerobic organisms is utilized by mitochondrial oxidases to produce adenosine triphosphate in a process coupled to the reduction of cellular O2 to water. About 1–4% of the O2 used in these reactions is converted to O2− and H2O2, which can be detrimental to mitochondrial function if not adequately detoxified.13 Cardiomyocytes contain the greatest density of mitochondria in the body, due to the energy requirements of the heart. Cardiac mitochondria in animal models of HF produce more O2− than normal mitochondria.14
Nox are enzyme complexes that catalyze O2 reduction using reduced nicotinamide adenine dinucleotide phosphate (NADPH) as the electron donor. This process produces ROS such as O2− and H2O2.17 These enzymes were originally identified in neutrophils. The structure of Nox in these cells includes a membrane complex made up of the catalytic subunit, gp91phox and the subunit p22phox, the cytoplasmic proteins p47phox, p67phox and p40phox, and Rac, a low molecular weight G protein.17,18 Several homologs of the gp91phox subunit, now known as Nox2, have been identified, and there are seven Nox isoforms, Nox1–5 and the dual oxidases Duox1 and Duox2, which differ in expression, molecular composition, subcellular location and tissue distribution.7,19 Nox1–3 require cytosolic subunits to be translocated to form a membrane complex in order to produce ROS,17 while Nox4 is constitutively active and Nox5 remains inactive until stimulated by calcium.11,17 The most important Nox in CVD are Nox1, Nox2, Nox4 and Nox5.20 The activity of Nox1 and Nox2 can be increased by angiotensin II, growth hormones and cytokines.11,17 In terms of distribution, endothelial cells express Nox2, Nox4 and Nox5, vascular smooth muscle cells express Nox1, Nox4 and Nox5, phagocytes mainly express Nox2,11 and cardiomyocytes mainly produce Nox2 and Nox4.19 The Nox family has been implicated in the pathogenesis of HF due to pressure overload, doxorubicin-induced HF and ischemic and diabetic cardiomyopathy.19
eNOS synthesizes NO and citrulline from O2 and l-arginine. However, in conditions of oxidative stress or reduced availability of the substrate (l-arginine) or the cofactor tetrahydrobiopterin (BH4), eNOS can become uncoupled and produce O2− instead of NO (Figure 3).21 In the heart, eNOS is expressed in endothelial cells and cardiomyocytes. Uncoupling of eNOS appears to contribute to endothelial dysfunction in hypertensive diastolic HF and to ventricular hypertrophy due to pressure overload.14,22
 eNOS requires the presence of a cofactor,
eNOS requires the presence of a cofactor, Effects of oxidative stress on endothelial nitric oxide synthase.
The synthesis of NO by eNOS requires the presence of a cofactor, BH4. When BH4 binds to eNOS, the enzyme is coupled. Degradation of BH4 by ROS, particularly ONOO− leads to the uncoupling of the enzyme and hence the production of O2− instead of NO. In turn, O2− reacts with NO, producing ONOO− and oxidizing BH4, leading to a vicious cycle of eNOS uncoupling. Akt: protein kinase B; AMPK: adenosine monophosphate-activated protein kinase; mRNA: messenger ribonucleic acid; BH4: tetrahydrobiopterin; Poly(A): polyadenylate; Cav-1: caveolin-1; eNOS: endothelial nitric oxide synthase; GTPCH-1: guanosine triphosphate cyclohydrolase; NO: nitric oxide; O2: oxygen; O2−: superoxide; ONOO−: peroxynitrite; P: phosphate; PI3K: phosphoinositide 3-kinase; PKA: protein kinase A; ROS: reactive oxygen species; Ser633: serine 633; Ser1177: serine 1177.
XO is involved in purine metabolism, catalyzing the conversion of hypoxanthine to xanthine and thence to uric acid. O2 is used as an electron donor in these reactions, producing O2− and H2O2.13 Increased expression and activity of XO has been described in HF, while administration of allopurinol, an XO inhibitor, increased cardiac contractility in an animal model of HF and reduced adverse cardiac remodeling in an experimental model of myocardial infarction (MI).14
The MAO family of enzymes catalyzes the oxidative degradation of neutrotransmitters such as noradrenaline, adrenaline and dopamine, and in the process generates H2O2. When the heart is subjected to neurohormonal and/or chronic hemodynamic stress, the abundance of circulating/tissue monoamines can increase MAO-derived H2O2 production,15 with significant contributions from both isoforms. MAO-A may establish a link with the renin-angiotensin system in the pathophysiology of diabetic cardiomyopathy,23 and there is evidence of cardiomyocyte necrosis when it is over-expressed,24 while MAO-B predominates in chronic hemodynamic overload.25
MPO is an enzyme secreted by activated neutrophils and monocytes in inflammatory conditions and uses H2O2 to produce several oxidizing molecules, including HClO, chloramines and nitrogen dioxides, that cause tissue damage in the heart and blood vessels and contribute to endothelial dysfunction.13 Plasma MPO is elevated in HF patients compared to controls, and its activity is increased in severe chronic HF compared to mild to moderate HF.16,26
Oxidative stress can result not only from increased ROS production but also from dysfunctional antioxidant defenses.21 The most important antioxidant enzymes are superoxide dismutase (SOD), catalase and glutathione peroxidase (GPx).13 Conversion of O2− to H2O2 is catalyzed by SOD isoforms found in the cytoplasm and organelles (Cu,Zn-SOD and SOD-1), mitochondria (SOD-2)' alt='manganese-dependent superoxide dismutase (mitochondrial isoform; SOD-2)'>MnSOD
and SOD-2) or extracellularly (extracellular superoxide dismutase [EC-SOD] or SOD-3). H2O2 can then be converted to H2O and O2 by catalase in peroxisomes or by GPx in the cytoplasm and mitochondria.13 Other antioxidant enzymes include glutathione reductase, glutathione-S-transferase, the peroxiredoxins and the thioredoxins. The latter regulate the thiol-disulfide status of proteins, affecting their structure and function, and thioredoxin 1 binds directly to and metabolize ROS.27 Heme oxygenase 1 (HO-1) also has antioxidant properties; as ROS formation can be catalyzed by an excess of heme, degradation of this compound by HO-1 reduces oxidative stress.28 The human body also has non-enzyme antioxidant defenses, including glutathione (GSH), vitamins C and E, carotenoids, uric acid, bilirubin and albumin.13 High-density lipoprotein (HDL) is another antioxidant molecule; functional HDL has high levels of antioxidant and anti-inflammatory proteins and enzymes, notably paraoxonase-1, which appears to be involved in inhibition of low-density lipoprotein (LDL) oxidation.29Although reduced antioxidant enzyme activity has been reported in animal models of HF,30 there are conflicting results in HF patients, with increases, decreases, and no change in antioxidant enzyme activity.31–33 Reductions in vitamin C and carotenoid levels have also been seen in patients with chronic HF.34
ROS are involved in several processes that affect cardiac structure and function, contributing to the genesis and progression of HF.
Reactive oxygen species and cardiac contractilityIn the heart, ROS can target the function of various ion channels (L-type calcium, sodium and potassium channels) and depress the activity of the sarcoplasmic reticulum Ca2+ ATPase SERCA2.35 They may also modify proteins that are important in cardiac contractility; for example, the phosphorylation of troponin T by ROS-activated kinases may contribute to reduced cardiac contractility.35
Reactive oxygen species and cardiac hypertrophy and fibrosisROS stimulate various enzymes of the mitogen-activated protein kinase (MAPK) family, including the extracellular signal-regulated kinases (ERK 1/2) and apoptosis signal-regulating kinase 1 (ASK1), contributing to the development of cardiac hypertrophy.35 Activation of transcription factors such as nuclear factor kappa B and activator protein 1 (AP-1) is also involved in ROS-induced cardiac hypertrophy.35 ROS can also stimulate proliferation of cardiac fibroblasts and activate extracellular matrix metalloproteinases (MMPs), leading to remodeling of the extracellular matrix. These alterations are important factors in adverse myocardial remodeling.14
Reactive oxygen species and cardiomyocyte apoptosisROS can mediate cardiomyocyte apoptosis by a variety of mechanisms, including direct genotoxicity, activation of ASK-1 in response to tumor necrosis factor alpha, and stimulation of kinases that induce mitochondrial death in response to activation of beta-adrenergic receptors.35 Interestingly, induction of cardiomyocyte hypertrophy or apoptosis appears to depend on the level of ROS produced: relatively low levels of H2O2 stimulate protein synthesis, while higher levels induce cardiomyocyte apoptosis.35
Reactive oxygen species and mitochondrial dysfunctionMitochondria are important sources of ROS in the heart, but they can also be affected by their toxic effects. ROS can damage mitochondrial DNA, reducing transcription and protein synthesis, leading to mitochondrial dysfunction, disruption of cardiac energy metabolism and cell death.14
Reactive oxygen species and ischemic cardiomyopathyROS can contribute to the genesis and progression of CAD. ROS produced in the vessel wall are involved in the formation of oxidized LDL (oxLDL), which is central to the pathogenesis of atherosclerosis.35 ROS-associated activation of MMPs may also play an important role in instability and rupture of atheromatous plaque in the coronary arteries, leading to thrombosis.35 ROS are also involved in reperfusion injury and tissue necrosis caused by MI.35
Interaction between reactive oxygen species and reactive nitrogen speciesThe interaction between ROS and NO can affect cardiac function. NO mediates protein S-nitrosylation at specific cysteine residues, which influences calcium flux and excitation-contraction coupling. High levels of O2− can inhibit protein S-nitrosylation, while the interaction of O2− with NO, as well as contributing to endothelial dysfunction by reducing NO availability, also produces peroxynitrite, a potent oxidant that induces apoptosis or cell necrosis.35,36
Statins and oxidative stressEffects of statins on endothelial nitric oxide synthaseeNOS is the main enzymatic source of NO in blood vessels. NO plays a part in vascular homeostasis, by inhibiting platelet activation and aggregation, vascular smooth muscle cell proliferation, expression of cell adhesion molecules and production of extracellular matrix.37 NO also helps regulate cardiac contractility and heart rate, limits cardiac remodeling after MI, and contributes to the protective effect of ischemic pre- and postconditioning.38
Binding to BH4 is essential for eNOS to synthesize NO. Degradation of this cofactor by ROS leads to uncoupling of the enzyme and production of O2− instead of NO, as mentioned previously11 (Figure 3). De novo synthesis of BH4 requires the enzyme guanosine triphosphate cyclohydrolase (GTPCH), a limiting factor for its formation. In human endothelial cells, fluvastatin and cerivastatin significantly increased the expression of messenger ribonucleic acid (mRNA) of GTPCH and intracellular BH4 concentrations.39,40 These statins also increased eNOS transcription.39,40 In another study, atorvastatin, pravastatin or pitavastatin also increased eNOS expression through stimulation of protein kinase B (Akt) at Ser473, inhibiting endothelial senescence induced by oxidative stress.41
Increased eNOS expression can also result from enhanced mRNA stability due to polyadenylation. Simvastatin and rosuvastatin significantly increased polyadenylation and hence the stability of eNOS mRNA in bovine aortic endothelial cells42 (Figure 4).
 eNOS and by preventing
eNOS and by preventing Action of statins on endothelial nitric oxide synthase.
Statins stimulate the synthesis of NO by eNOS and by preventing eNOS uncoupling, limit the formation of ROS. Akt: protein kinase B; AMPK: adenosine monophosphate-activated protein kinase; mRNA: messenger ribonucleic acid; BH4: tetrahydrobiopterin; eNOS: endothelial nitric oxide synthase; Poly(A): polyadenylate; Cav-1: caveolin-1; GTPCH-1: guanosine triphosphate cyclohydrolase; NO: nitric oxide; O2: oxygen; P: phosphate; PI3K: phosphoinositide 3-kinase; PKA: protein kinase A; Poly(A): polyadenylate; ROS: reactive oxygen species; Ser633: serine 633; Ser1177: serine 1177.
Statins can also contribute to increased eNOS activity through phosphorylation. Fluvastatin and pitavastatin promote eNOS phosphorylation at Ser1177 and/or Ser633 via phosphoinositide 3-kinase (PI3K)/Akt and protein kinase A (PKA), respectively, increasing eNOS activity in human endothelial cells.39,43 When adenosine monophosphate-activated protein kinase (AMPK) is stimulated by statins, it can also phosphorylate eNOS at Ser1177, increasing its activity.44 Statins can also enhance eNOS activity by inhibiting the expression of caveolin-1,45 which interacts with eNOS in endothelial cells and reduces its activity.46 Furthermore, they can increase eNOS activity and coupling by reducing concentrations of asymmetric dimethylarginine, an eNOS inhibitor.11
Effects of statins on nicotinamide adenine dinucleotide phosphate oxidasesAs stated above, Nox are important sources of ROS and contribute to oxidative stress. Production of ROS by the isoforms Nox2 and Nox1 requires the activation and translocation of cytoplasmic proteins such as Rac, p47phox or its homolog NoxO1, and p67phox or its homolog NoxA1, which then interact with the membrane subunits Nox1 or gp91phox (Nox2) and p22phox. The subunit p40phox is also translocated in Nox2. Activation and translocation of the protein Rac is dependent on isoprenylation by GGPP, which is synthesized via the mevalonate pathway.11 Inhibition of this pathway by statins blocks Rac activation and thereby reduces Nox1 and Nox2 activity11 (Figure 5).
 NADPH oxidases are composed of membrane subunits such as p22phox and gp91phox (Nox2), and cytosolic subunits including p47phox, p67phox, p40phox and
NADPH oxidases are composed of membrane subunits such as p22phox and gp91phox (Nox2), and cytosolic subunits including p47phox, p67phox, p40phox and Action of statins on nicotinamide adenine dinucleotide phosphate oxidase in endothelial cells.
NADPH oxidases are composed of membrane subunits such as p22phox and gp91phox (Nox2), and cytosolic subunits including p47phox, p67phox, p40phox and GTP-Rac. Activation of Nox requires activation and membrane translocation of Rac and p47phox, p67phox and p40phox, while activation of Rac involves isoprenylation by GGPF. Inhibition of the formation of GGPF by statins prevents activation of Rac and hence of Nox2. Another mechanism by which statins appear to inhibit Nox2 is by altering the phosphorylation of PKC and the membrane translocation of p47phox and is partly mediated by adiponectin, which inhibits p47phox translocation. ADP: adenosine diphosphate; FPF: farnesyl pyrophosphate; GDP: guanosine diphosphate; GGPP: geranylgeranyl pyrophosphate; GTP: guanosine triphosphate; HMG-CoA: hydroxymethylglutaryl-coenzyme A; mRNA: messenger ribonucleic acid; Nox: NADPH oxidase; O2: oxygen; O2−: superoxide; PKC: protein kinase C.
Statins’ inhibitory effect on Nox activity appears to affect other subunits of these enzymes. Reduced mRNA expression of p22phox, gp91phox and Nox1, protein expression of p47phox and translocation of p47phox and p67phox have been observed following statin therapy; they also inhibit the expression of the angiotensin II receptor AT1, which mediates the stimulating effect of angiotensin II on Nox activity.11
It has recently been suggested that statins’ inhibition of Nox may be partly mediated by adiponectin, a protein synthesized by adipocytes. In hypercholesterolemic patients, increases in adiponectin levels following treatment with atorvastatin were associated with decreases in the soluble form of gp91phox, platelet ROS production and urinary isoprostanes. In-vitro treatment with adiponectin also inhibited p47phox translocation and soluble gp91phox cleavage, inhibiting Nox activation in platelets.47
Effects of statins on antioxidant systemsStatins can stimulate antioxidant defenses. In-vitro treatment of human endothelial cells with atorvastatin increased expression of catalase and SOD-2)' alt='manganese-dependent superoxide dismutase (mitochondrial isoform; SOD-2)'>MnSOD
following phosphorylation of Akt at Ser473.41 In tumor cells, fluvastatin increased mRNA and protein expression of SOD-2)' alt='manganese-dependent superoxide dismutase (mitochondrial isoform; SOD-2)'>MnSOD and caused a two-fold increase in SOD-2)' alt='manganese-dependent superoxide dismutase (mitochondrial isoform; SOD-2)'>MnSOD activity.48 These effects appear to be due to downregulation of DNA damage-binding protein 2 (DDB2), a negative regulator of SOD-2)' alt='manganese-dependent superoxide dismutase (mitochondrial isoform; SOD-2)'>MnSOD transcription.48 Fluvastatin also increased catalase activity and GSH concentrations.48 Increased GSH has also been observed in promyelocytic cells treated with rosuvastatin, and appears to result from upregulation of gamma-glutamylcysteine synthetase, the rate-limiting enzyme of GSH.49Statins also influence other antioxidant enzymes, such as thioredoxin 1 and HO-1. In human endothelial cells, atorvastatin stimulated NO synthesis and hence S-nitrosylation of thioredoxin 1, increasing its activity and reducing intracellular ROS,27 while rosuvastatin increased mRNA and protein expression of HO-1.50
Statins may have a further protective effect through their action on PON1, an enzyme involved in the antioxidant effect of HDL, the activity of which was increased by atorvastatin in hypercholesterolemic individuals.51
Other antioxidant/anti-inflammatory effects of statinsThe relationship between oxidative stress and inflammation has been demonstrated in HF and other chronic diseases, in which it contributes to their genesis and progression.52,53 Oxidative stress activates various transcription factors, including NF-κB and AP-1, that induce the expression of proinflammatory cytokines, chemokines and adhesion molecules.53,54 At the same time, the production of large quantities of ROS is a characteristic of activated inflammatory cells.53 Some cytokines also trigger ROS production in vascular endothelial and smooth muscle cells.55,56
Statins affect various molecules that play an important role in both oxidative stress and inflammation. For example, MPO, which is found in neutrophils and monocytes, contributes to ROS formation during the inflammatory process, thereby triggering lipid oxidation and tissue damage.57 MPO interferes with endothelial homeostasis, contributing to the onset and progression of atherosclerosis,57 and is also associated with worsening New York Heart Association (NYHA) functional class.16 There is evidence that statins strongly inhibit MPO mRNA expression in human and murine monocyte-macrophages, an effect dependent on blockade of the mevalonate pathway and hence reduction the formation of GGPP formation.58
Galectin-3, a member of the lectin family of proteins, is another biomarker associated with oxidative stress and inflammation. It is involved in proliferation, chemotaxis, phagocytosis, apoptosis, angiogenesis and myocardial fibrosis, all of which are mechanisms underlying the pathogenesis of cardiovascular disease (CVD). The release of galectin-3 by monocytes and macrophages is regulated by ROS and Nox. Curiously, in vitro, galectin-3 stimulates synthesis of O2− by monocytes and macrophages, and so it may contribute to the vicious cycle of oxidative stress and inflammation.59 In an animal model of atherosclerosis, galectin-3 expression increased in proportion to the degree of plaque extent and inflammation, and atorvastatin treatment markedly reduced intraplaque galectin-3 and macrophage signals.60
It has recently been shown that statins reduce inflammation by stimulating the synthesis of 15-epi-lipoxin A4 (15-epi-LXA4), which has pro-resolving, anti-inflammatory, antioxidant, vasodilatory and antiproliferative properties61–66 (Figure 6). Synthesis of 15-epi-LXA4 from arachidonic acid involves the sequential action of cyclooxygenase 2 (COX-2) and 5-lipoxygenase (5-LOX). Statins induce expression and S-nitrosylation of COX-2, resulting in the synthesis of 15R-hydroxyeicosatetraenoic acid, which is then converted via 5-LOX to 15-epi-LXA4 (Figure 6). Treatment with atorvastatin significantly increased the synthesis of 15-epi-LXA4 in rat myocardium via S-nitrosylation of COX-2.62 In an animal model of airway inflammation, treatment with lovastatin also induced the formation of 15-epi-LXA4 and markedly reduced acute lung inflammation. Furthermore, in-vitro lovastatin increased 15-epi-LXA4 production during interactions between polymorphonuclear cells and human airway epithelial cells primed with cytokines63 (Figure 7).
 COX-2 by statins promotes the synthesis of 15-epi-lipoxins, which have protective effects in various types of cell and help reduce inflammation. ↑: increased; ↓: decreased; AA: arachidonic acid; CCR5: C-C chemokine receptor type 5; CR3: complement receptor 3 (CD11B, cluster of differentiation 11B/CD18, integrin beta-2);
COX-2 by statins promotes the synthesis of 15-epi-lipoxins, which have protective effects in various types of cell and help reduce inflammation. ↑: increased; ↓: decreased; AA: arachidonic acid; CCR5: C-C chemokine receptor type 5; CR3: complement receptor 3 (CD11B, cluster of differentiation 11B/CD18, integrin beta-2); Statins and synthesis of 15-epi-lipoxins: protective effects.
S-nitrosylation of COX-2 by statins promotes the synthesis of 15-epi-lipoxins, which have protective effects in various types of cell and help reduce inflammation. ↑: increased; ↓: decreased; AA: arachidonic acid; CCR5: C-C chemokine receptor type 5; CR3: complement receptor 3 (CD11B, cluster of differentiation 11B/CD18, integrin beta-2); COX-2: cyclooxygenase-2; HO-1: heme oxygenase-1; ICAM-1: intercellular adhesion molecule 1; MMP-3: matrix metalloproteinase-3; NO: nitric oxide; O2−: superoxide; ONOO− peroxynitrite; PGI2: prostacyclin; ROS: reactive oxygen species; TNF-α: tumor necrosis factor alpha; VEFG: vascular endothelial growth factor; 5-LOX: 5-lipoxygenase; 15R-HETE: 15-hydroxyicosatetraenoic acid; 15-epi-LXA4: 15-epi-lipoxin A4.
 BH4: tetrahydrobiopterin;
BH4: tetrahydrobiopterin; Summary of statins’ antioxidant and anti-inflammatory effects. ↑: increased; ↓: decreased; ADMA: asymmetric dimethylarginine; BH4: tetrahydrobiopterin; eNOS: endothelial nitric oxide synthase; GSH: glutathione; GTPCH: guanosine triphosphate cyclohydrolase; HDL: high-density lipoprotein; HO-1: heme oxygenase-1; SOD-2)' alt='manganese-dependent superoxide dismutase (mitochondrial isoform; SOD-2)'>MnSOD: manganese-dependent superoxide dismutase; MPO: myeloperoxidase; mRNA: messenger ribonucleic acid; Nox: nicotinamide adenine dinucleotide phosphate oxidase; PON-1: paraoxonase-1; 15-epi-LXA4: 15-epi-lipoxinA4.
Oxidative stress is associated with aging and the development of many diseases, including CVD.14 Several studies over the last 10 years have assessed the effects of statins on markers of oxidative stress and endothelial function in cardiovascular, renal and metabolic disorders.
In HF patients with reduced left ventricular ejection fraction (LVEF), one month of simvastatin or atorvastatin therapy reduced ROS production and systemic concentrations of malondialdehyde (MDA), a marker of lipid peroxidation, increased EC-SOD activity, and improved endothelial function and functional capacity.67,68 In another study, of patients with systolic HF, one month's treatment with rosuvastatin also significantly reduced plasma MPO and oxLDL levels.69
In individuals with CAD undergoing coronary artery bypass grafting, prospective treatment with atorvastatin or pravastatin for four weeks significantly reduced mRNA expression and activity of Rac1 in myocardial tissue samples, as well as angiotensin II-induced Nox activity.70 In a study of patients undergoing cardiac surgery who were monitored until discharge, a strong association was observed between O2− and peroxynitrite production and postoperative complications during hospital stay. Preoperative atorvastatin therapy for three days significantly reduced Nox activity and O2− and peroxynitrite production in myocardial tissue, while ex-vivo incubation of myocardium with atorvastatin induced mevalonate-reversible and Rac1-mediated inhibition of Nox.71 A similar study showed that preoperative administration of atorvastatin increased BH4 availability and reduced both basal and uncoupled eNOS-derived O2− production in samples of internal mammary artery.72 Statin therapy also appears to reduce plasma MPO concentrations in patients with acute coronary syndrome but not in those with stable CAD.73
Statins’ redox effects have been assessed in individuals with hyperlipidemia, in whom they have various protective effects, including reduced ROS production and increased platelet NO synthesis, decreased urinary and platelet isoprostanes, diminished platelet Rac1 activation, inhibiting of serum and platelet Nox2, increasing adiponectin levels and thereby reducing gp91phox and Nox activity, raised serum vitamin E levels, and decreased DNA damage in hypercholesterolemic individuals with the C242T polymorphism of the p22phox gene, which is associated with risk for developing CAD.47,74–78
Oxidative stress is also linked to the pathogenesis of atrial fibrillation (AF). In a study assessing the effects of statins on ROS production in patients who developed AF following cardiac surgery or with permanent AF, atorvastatin reduced Rac1 and Nox activity in right atrial samples from those who developed postoperative AFF, but did not affect ROS, eNOS uncoupling, or BH4 in those with permanent AF. These results suggest that Nox upregulation is an early but transient event in the natural history of AF.79
Statins also appear to have a protective effect in diabetes. Rosuvastatin treatment for six months in patients with diabetic nephropathy improved renal function and significantly reduced serum lipid peroxidation products and urinary 8-hydroxy-2′-deoxyguanosine (8-OHdG), a marker of DNA oxidation.80 In diabetic polyneuropathy, rosuvastatin for 12 months led to improvements in severity, symptoms and nerve conduction parameters, as well as reductions in lipid peroxidation.81
Despite the growing evidence of statins’ antioxidant effects in cardiovascular and metabolic disorders, these effects are not seen in some studies. In patients at high risk for cardiovascular disease, atorvastatin and simvastatin did not significantly alter plasma MDA or MPO concentrations or urinary 8-OHdG,82 while in another study of patients with diabetic nephropathy, atorvastatin did not increase NO availability.83
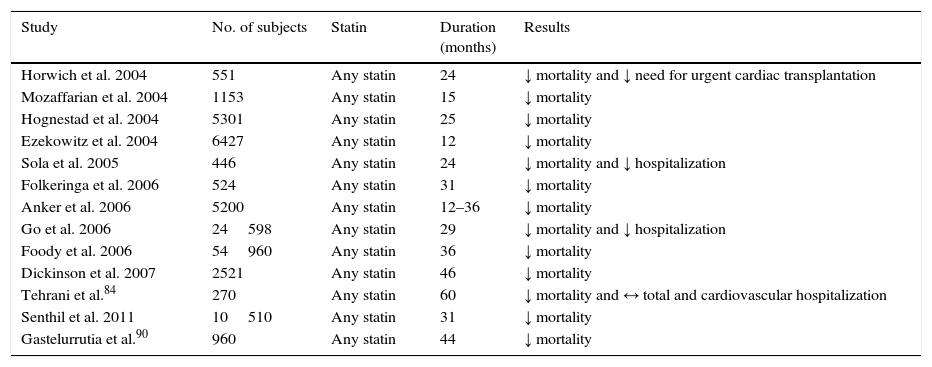
Statins in chronic heart failure: clinical trialsMost studies on the effects of statins in HF are observational and non-randomized, and focus on HF with reduced LVEF (≤35%). They suggest that statins reduce mortality (Table 2); a reduction in mortality has also been reported in HF with preserved LVEF after treatment with statins.84
Effects of statins in heart failure: non-randomized observational studies (adapted from6–8,84,90).
| Study | No. of subjects | Statin | Duration (months) | Results |
|---|---|---|---|---|
| Horwich et al. 2004 | 551 | Any statin | 24 | ↓ mortality and ↓ need for urgent cardiac transplantation |
| Mozaffarian et al. 2004 | 1153 | Any statin | 15 | ↓ mortality |
| Hognestad et al. 2004 | 5301 | Any statin | 25 | ↓ mortality |
| Ezekowitz et al. 2004 | 6427 | Any statin | 12 | ↓ mortality |
| Sola et al. 2005 | 446 | Any statin | 24 | ↓ mortality and ↓ hospitalization |
| Folkeringa et al. 2006 | 524 | Any statin | 31 | ↓ mortality |
| Anker et al. 2006 | 5200 | Any statin | 12–36 | ↓ mortality |
| Go et al. 2006 | 24598 | Any statin | 29 | ↓ mortality and ↓ hospitalization |
| Foody et al. 2006 | 54960 | Any statin | 36 | ↓ mortality |
| Dickinson et al. 2007 | 2521 | Any statin | 46 | ↓ mortality |
| Tehrani et al.84 | 270 | Any statin | 60 | ↓ mortality and ↔ total and cardiovascular hospitalization |
| Senthil et al. 2011 | 10510 | Any statin | 31 | ↓ mortality |
| Gastelurrutia et al.90 | 960 | Any statin | 44 | ↓ mortality |
↓: decreased; ↓: increased; ↔: no change.
However, there have been some randomized clinical trials on statins in HF, the largest being Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico-Heart Failure (GISSI-HF)85 and Controlled Rosuvastatin Multinational Trial in Heart Failure (CORONA).86
CORONA was a prospective randomized clinical trial of rosuvastatin use in 5011 patients with symptomatic HF (NYHA II–IV) of ischemic etiology, with LVEF ≤40% (NYHA III or IV) or ≤35% (NYHA II) and aged ≥60 years.86 Despite improvements in lipid profile and reduced high-sensitivity C-reactive protein, there were no significant differences between the two groups in cardiovascular mortality, MI or stroke.87
GISSI-HF was a prospective randomized trial of 4574 patients aged ≥18 years with HF in NYHA II–IV, irrespective of cause and LVEF, randomized to rosuvastatin or placebo. Although daily rosuvastatin was safe, it did not affect clinical outcomes (time to death or admission to hospital for cardiovascular reasons) during a median follow-up of 46 months.85
Various explanations have been put forward for the results of CORONA and GISSI-HF. It is possible that not all statins have the same effects, since atorvastatin, unlike rosuvastatin, reduces overall mortality and hospitalizations, improves LVEF88 and reduces systemic B-type natriuretic peptide (BNP), which may be due to differences in the pharmacokinetic properties of different statins.89 Furthermore, a meta-analysis found no correlation between dose and results, suggesting that the type of statin may be more important than the dose.88
The characteristics of the study populations, particularly age (mean age in CORONA and GISSI-HF was 73 and 68 years, respectively) and the exclusion of patients previously medicated with statins, may also have affected the results.90
According to another theory, statins may be beneficial in the early stages of HF but do not prevent progressive deterioration of cardiac function in more advanced stages.91 It would thus be useful to identify patient subgroups who would derive greater benefit from these drugs. Patients treated with rosuvastatin and in the lower tertile (<103 pmol/l or <868 pg/ml) of N-terminal brain-type natriuretic peptide (NT-proBNP) appear to have greater reductions in cardiovascular mortality, MI and stroke.92 Similarly, patients treated with rosuvastatin and with galectin-3 ≤19 ng/ml presented lower rates of primary events, total mortality and HF hospitalizations than placebo; those with higher galectin-3 levels did not present similar benefits.93
The European Society of Cardiology (ESC) and the American College of Cardiology (ACC) do not currently recommend statins as adjuvant therapy in HF in the absence of other indications for their use.2,3
ConclusionsIn addition to their effects on cholesterol synthesis, statins have pleiotropic properties, notably antioxidant action. These effects appear to result from inhibition of the synthesis of isoprenoid intermediates of the mevalonate pathway.
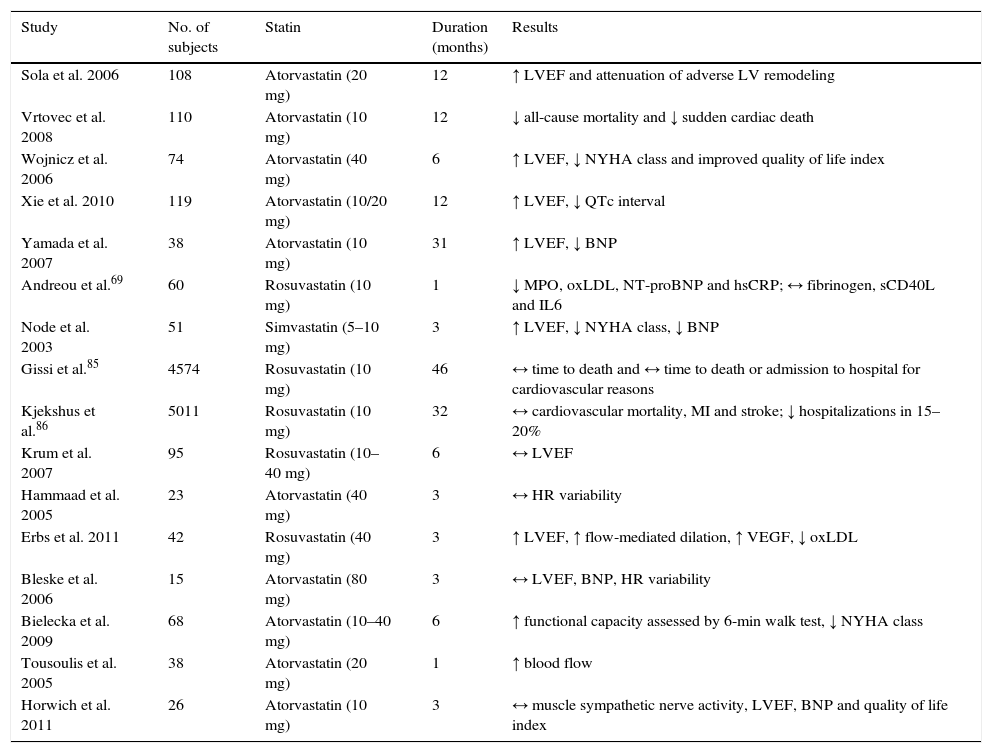
There is widespread recognition of the role of oxidative stress in the pathogenesis of many diseases, including HF. ROS interfere with various processes that affect cardiac structure and function, contributing to apoptosis and mitochondrial dysfunction in cardiomyocytes, contractile dysfunction, myocardial fibrosis and hypertrophy, endothelial dysfunction and atherosclerosis. Statins reduce ROS levels and increase NO synthesis, thereby improving cardiac redox balance. These effects are mainly mediated by inhibition of pro-oxidant enzymes such as Nox and by stimulation of the expression, activity and stability of eNOS. In addition, statins stimulate other antioxidant enzymes and help control inflammatory processes. Statins thus have effects on various molecules that are central to both oxidative stress and inflammation, notably by stimulating the synthesis of 15-epi-LXA4, which has pro-resolving, anti-inflammatory and antioxidant properties. Their antioxidant effects have been demonstrated in several cardiovascular conditions, including HF, CAD and AF, mainly in the early stages of these diseases (Table 3).
Effects of statins in heart failure with reduced ejection fraction: non-randomized studies (adapted from6–8,69,85,87).
| Study | No. of subjects | Statin | Duration (months) | Results |
|---|---|---|---|---|
| Sola et al. 2006 | 108 | Atorvastatin (20 mg) | 12 | ↑ LVEF and attenuation of adverse LV remodeling |
| Vrtovec et al. 2008 | 110 | Atorvastatin (10 mg) | 12 | ↓ all-cause mortality and ↓ sudden cardiac death |
| Wojnicz et al. 2006 | 74 | Atorvastatin (40 mg) | 6 | ↑ LVEF, ↓ NYHA class and improved quality of life index |
| Xie et al. 2010 | 119 | Atorvastatin (10/20 mg) | 12 | ↑ LVEF, ↓ QTc interval |
| Yamada et al. 2007 | 38 | Atorvastatin (10 mg) | 31 | ↑ LVEF, ↓ BNP |
| Andreou et al.69 | 60 | Rosuvastatin (10 mg) | 1 | ↓ MPO, oxLDL, NT-proBNP and hsCRP; ↔ fibrinogen, sCD40L and IL6 |
| Node et al. 2003 | 51 | Simvastatin (5–10 mg) | 3 | ↑ LVEF, ↓ NYHA class, ↓ BNP |
| Gissi et al.85 | 4574 | Rosuvastatin (10 mg) | 46 | ↔ time to death and ↔ time to death or admission to hospital for cardiovascular reasons |
| Kjekshus et al.86 | 5011 | Rosuvastatin (10 mg) | 32 | ↔ cardiovascular mortality, MI and stroke; ↓ hospitalizations in 15–20% |
| Krum et al. 2007 | 95 | Rosuvastatin (10–40 mg) | 6 | ↔ LVEF |
| Hammaad et al. 2005 | 23 | Atorvastatin (40 mg) | 3 | ↔ HR variability |
| Erbs et al. 2011 | 42 | Rosuvastatin (40 mg) | 3 | ↑ LVEF, ↑ flow-mediated dilation, ↑ VEGF, ↓ oxLDL |
| Bleske et al. 2006 | 15 | Atorvastatin (80 mg) | 3 | ↔ LVEF, BNP, HR variability |
| Bielecka et al. 2009 | 68 | Atorvastatin (10–40 mg) | 6 | ↑ functional capacity assessed by 6-min walk test, ↓ NYHA class |
| Tousoulis et al. 2005 | 38 | Atorvastatin (20 mg) | 1 | ↑ blood flow |
| Horwich et al. 2011 | 26 | Atorvastatin (10 mg) | 3 | ↔ muscle sympathetic nerve activity, LVEF, BNP and quality of life index |
↓: decreased; ↓: increased; ↔: no change; BNP: B-type natriuretic peptide; MI: myocardial infarction; HR: heart rate; LVEF: left ventricular ejection fraction; hsCRP: high-sensitivity C-reactive protein; IL6: interleukin 6; LV: left ventricular; MPO: myeloperoxidase: NYHA: New York Heart Association; NT-proBNP: N-terminal of B-type natriuretic peptide; oxLDL: oxidized low-density lipoprotein; QTc: corrected QT interval; sCD40L: soluble CD40 ligand; VEGF: vascular endothelial growth factor.
With regard to the effects of statins in reducing cardiovascular events and mortality in HF, although various observational studies have shown reduced mortality with statin therapy, two large clinical trials – CORONA and GISSI-HF – did not demonstrate such a benefit. The ESC and the ACC do not currently recommend statins in HF in the absence of other indications for their use. Given the lack of consensus, further clinical trials are needed to clarify the clinical value of statins in subgroups of HF patients and its relation with their effects on redox state.
FundingT. Sousa acknowledges support from Fundação para a Ciência e a Tecnologia (FCT) (SFRH/BPD/112005/2015).
Ethical responsibilitiesProtection of people and animalsThe authors declare that no experiments were performed on humans or animals for this study.
Data confidentialityThe authors declare that no patient data appear in this article.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflicts of interestThe authors have no conflicts of interest to declare.

www.publicationethics.org.