






A miocardiopatia hipertrófica (MCH) é uma patologia com transmissão essencialmente autossómica dominante, expressão clínica variável e penetrância incompleta. O rastreio familiar tem por objetivo identificar a ocorrência ou o risco de desenvolvimento da doença nos parentes em primeiro grau do caso índex. As normas de orientação da ESC e da ACCF/AHA recomendam a avaliação dos familiares em idade pediátrica a partir dos 10‐12 anos.
ObjetivosAvaliaram‐se os resultados de um programa de rastreio pediátrico de MCH familiar e o valor preditivo do estudo genético nexte contexto. Foi ainda aferida a penetrância fenotípica ao longo do tempo de seguimento destas crianças.
Métodos e resultadosForam incluídas 20 crianças pertencentes a dez famílias (2004‐2013). Três das crianças constituíram‐se como o caso índex, sendo as restantes parentes em primeiro grau de um doente com MCH (80% sexo masculino; idade mediana=10 anos). Catorze crianças eram portadoras de mutação de um gene sarcomérico (70%; idade mediana=8 anos). Sete (50%) dos 14 portadores de mutação apresentavam fenótipo positivo na primeira avaliação.
Foram definidos como «familiares em risco» aqueles com teste genético positivo, mas com fenótipo normal à apresentação. Após 3,5±0,8 anos de seguimento, duas das crianças fenótipo negativo portadoras de mutação (gene MYBPC3) desenvolveram MCH, aos dez e 15 anos de idade (28% de taxa de penetrância).
ConclusõesA penetrância de MCH em crianças com fenótipo normal à apresentação foi de 28% após 3,5 anos de seguimento. Tal sublinha a importância da avaliação longitudinal dos portadores de mutação de genes sarcoméricos, independentemente da presença de fenótipo patológico.
Hypertrophic cardiomyopathy (HCM) is most often of autosomal dominant inheritance with incomplete penetrance and variable expression. The main purpose of family screening is to identify relatives with unrecognized HCM and to monitor those at risk for disease, in order to minimize complications and to assess risk of sudden cardiac death. The ESC and ACCF/AHA guidelines on the diagnosis and management of HCM recommend the screening of child relatives from the age of 10‐12 years.
ObjectivesWe studied the outcome of clinical screening and genetic testing of child probands and relatives (<18 years of age) from families with HCM and assessed the age‐related penetrance of HCM during the follow‐up of these young relatives.
Methods and ResultsTwenty patients from ten families were included between 2004 and 2013, consisting of three probands and 17 first‐degree relatives (80% male; median age 10 years). Fourteen child relatives were mutation carriers (70%; median age eight years). Seven (50%) of the 14 mutation carriers were diagnosed with HCM at initial assessment. At‐risk child relatives were defined as those with a positive mutation but a negative phenotype at enrollment.
After 3.5±0.8 years of follow‐up, two of the phenotype‐negative mutation carriers developed HCM at 10 and 15 years of age (28% penetrance rate).
ConclusionsThe penetrance of HCM in phenotype‐negative child relatives was 28% after 3.5 years of follow‐up. This underlines the need for long‐term monitoring of mutation carriers irrespective of the presence of a positive phenotype.
Actin, alpha, cardiac muscle 1
Ácido desoxirribonucleico
American College of Cardiology and American Heart Association
Cardio‐desfibrilhador implantável
Eletrocardiograma
European Society of Cardiology
Human Gene Mutation Database
Miocardiopatia hipertrófica
Morte súbita cardíaca
Myosin binding protein C
Myosin heavy chain
Myosin light chain 2
Myosin light chain 3
Número
Polymerase chain reaction
Single nucleotide polymorphism
Single nucleotide polymorphism database
Superfície corporal
Systolic anterior movement
Cardiac troponin T
Troponin I Type 3
Tropomyosin 1 (Alpha)
Taquicardia ventricular
Trato de saída do ventrículo esquerdo
Ventrículo esquerdo
A miocardiopatia hipertrófica (MCH) é uma patologia de transmissão essencialmente autossómica dominante, expressão clínica variável e penetrância incompleta, dependente da idade1.
As manifestações clínicas desta patologia são igualmente heterogéneas, desde a ausência de sintomas até à apresentação com sinais francos de insuficiência cardíaca ou morte súbita2.
O objetivo do rastreio familiar é o de identificar a ocorrência ou o risco de desenvolvimento da doença nos parentes em primeiro grau do caso índex.
As mais recentes normas de orientação da European Society of Cardiology (ESC) e da American College of Cardiology and American Heart Association (ACCF/AHA) recomendam a avaliação dos familiares em idade pediátrica a partir dos 10‐12 anos3,4.
Estima‐se que em cerca de 50‐60% dos casos de MCH familiar seja possível identificar uma mutação nos genes que codificam as proteínas sarcoméricas5.
No entanto, em crianças com fenótipo negativo, o valor prognóstico da identificação de mutações de genes sarcoméricos permanece incerto.
Avaliaram‐se os resultados do rastreio pediátrico de MCH familiar num centro de referenciação terciária de cardiologia pediátrica e o valor preditivo do estudo genético neste contexto. Foi ainda aferida a penetrância fenotípica da doença, ao longo do tempo de seguimento destas crianças.
MétodosPopulação em estudoTodos os casos de MCH familiar foram seguidos num centro de referenciação terciária de cardiologia pediátrica, em consulta dedicada, entre 2004‐2013. Foram incluídas crianças com idade inferior a 18 anos, pertencentes a famílias em que o caso índex tinha estudo genético positivo para mutação de gene sarcomérico.
A referenciação dos descendentes em primeiro grau do caso índex diagnosticado com MCH foi feita, primariamente, através da consulta de cardiologia do mesmo centro.
Nos casos em que o caso índex era uma criança, os seus irmãos foram referenciados à consulta de cardiologia pediátrica e os pais à consulta de cardiologia.
Todos os doentes foram ainda referenciados à consulta de genética médica do mesmo centro, para aconselhamento genético.
Avaliação clínica e estudo genéticoA avaliação inicial desta população incluiu observação clínica, eletrocardiograma (ECG) de 12 derivações em repouso, ecocardiograma transtorácico e teste genético para as oito mutações de genes sarcoméricos mais frequentes (MYH7, MYL2, MYL3, MYBPC3, TNNI3, TNNT2, TPM1 e ACTC1).
A pesquisa de mutações nos genes MYH7, MYL2, MYL3, MYBPC3, TNNI3, TNNT2, TPM1 e ACTC1 (totalidade da região codificante, incluindo transições intrão‐exão) foi efetuada através da técnica de polymerase chain reaction (PCR), com sequenciação direta (combinação de sequenciação de nova geração com coverage mínima de 30X e sequenciação de Sanger) dos produtos de PCR. A metodologia utilizada tem uma sensibilidade analítica de 99% para a deteção de substituições nucleotídicas e pequenas deleções e inserções.
Para classificação das variantes de ADN quanto à sua patogenicidade foram consultadas as bases de dados da Clinvar e da Human Gene Mutation Database (HGMD). Nas situações em que a mutação não tinha sido previamente descrita e para as variantes genéticas de significado incerto, foram utilizadas as ferramentas bioinformáticas de predição de patogenicidade PolyPhen‐2 e Mutation Taster, para aferição da sua relevância funcional.
As crianças com mais do que uma mutação foram definidas como apresentando um genótipo complexo.
A avaliação ecocardiográfica bidimensional, em modo M e Doppler, foi realizada de acordo com as recomendações da American Society of Echocardiography6.
Foram avaliadas as dimensões das cavidades cardíacas, septo interventricular e parede posterior do ventrículo esquerdo (VE), pesquisado SAM da válvula mitral e gradiente na câmara de saída do VE, em repouso e durante a manobra de Valsava. Foi definido como obstrução do trato de saída do ventrículo esquerdo (TSVE) um gradiente em repouso≥30mmHg3.
Do ponto de vista ecocardiográfico, o diagnóstico de MCH foi feito quando o valor máximo de espessura da parede posterior do VE era superior a duas vezes o desvio padrão (DP) da média prevista, relativamente à superfície corporal (SC)3. A SC foi calculada de acordo com a fórmula de Haycock7.
O traçado eletrocardiográfico foi analisado quanto à presença de desvio do eixo do QRS, inversão da onda T>1mm, infra desnivelamento do segmento ST>2mm e onda S>onda R em V4. Foram calculados a soma da amplitude de QRS nas derivações dos membros, o produto da amplitude‐duração de QRS nas 12 derivações e o QTc (segundo a fórmula de Bazett)8.
A estratificação do risco de morte súbita foi realizada de acordo com o modelo descrito por Östman‐Smith et al.9. Os doentes foram pontuados nos oito parâmetros analisados de 1‐3 (score máximo=14). Um score≥6 representa um risco elevado de morte súbita cardíaca. Apesar deste modelo preditivo ter sido desenvolvido para adultos com MCH, o mesmo modelo foi aplicado por estes autores a uma população pediátrica, tendo obtido semelhante valor preditivo (estudo não publicado, apresentado no 46th Annual Meeting of the Association for European Paediatric and Congenital Cardiology, Istambul, Turquia, 23‐26 de maio 2012).
Foram ainda analisados os seguintes fatores de risco para morte súbita cardíaca10:
- (I)
história familiar de morte súbita cardíaca: morte não‐traumática e prematura (idade<40 anos); morte ocorrida uma hora após início dos sintomas na ausência de sintomatologia prévia, incluindo morte noturna inesperada e não presenciada ou equivalentes, como episódio de ressuscitação cardiorrespiratória ou choque apropriado de cardio‐desfibrilhador implantável (CDI);
- (II)
síncope inexplicada de etiologia não neurocardiogénica;
- (III)
taquicardia ventricular não mantida: um ou mais períodos de≥3 extrasístoles ventriculares consecutivas, a uma frequência>120bpm com duração<30 segundos, em prova de esforço ou em Holter de 24 horas;
- (IV)
hipertrofia ventricular esquerda extrema: espessura máxima da parede do VE≥30mm ou Z‐score≥63.
Os doentes com genótipo e fenótipo positivos foram definidos como familiares afetados e seguidos em consulta específica de cardiologia pediátrica, com uma periodicidade semestral.
Foram definidos como familiares em risco de desenvolvimento de MCH, portadores de mutação de gene sarcomérico, variantes genéticas de significado incerto ou mutações não anteriormente descritas como associadas à ocorrência de MCH, na ausência de manifestações fenotípicas de doença.
Num dos casos, o estudo genético encontra‐se pendente e foi considerado existir risco de desenvolvimento de doença. As crianças nestas condições foram seguidas com periodicidade anual.
As crianças com fenótipo negativo e ausência de mutação foram consideradas como não tendo risco de desenvolvimento de MCH e dispensadas de seguimento.
As consultas de seguimento incluíram avaliação clínica, realização de ECG de 12 derivações e ecocardiograma transtorácico. Foi requisitada monitorização Holter de 24 horas sempre que clinicamente relevante.
Todos os doentes com idade superior a sete anos realizaram prova de esforço convencional. Dois dos doentes foram adicionalmente submetidos a ecocardiograma de esforço.
Análise estatísticaAs variáveis contínuas são apresentadas como médias e desvio padrão quando obedecendo a distribuição normal e como medianas, mínimo e máximo quando não obedecendo a distribuição normal. As variáveis categóricas são descritas de acordo com a sua frequência e percentagem.
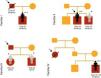
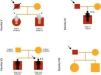
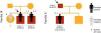
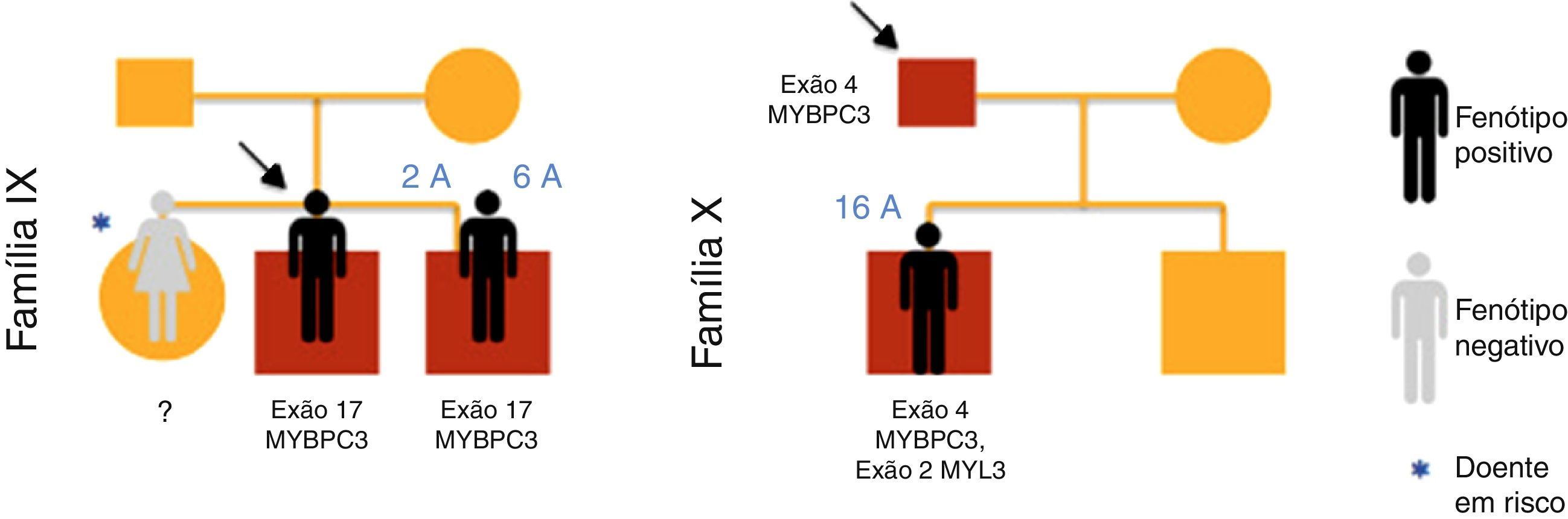
ResultadosForam incluídas no nosso estudo de MCH familiar 20 crianças pertencentes a 10 famílias (Figuras 1–3). Três das crianças constituíram‐se como o caso índex e as restantes eram parentes em primeiro grau de um doente com MCH (80% sexo masculino; idade mediana=10 anos; min=1 mês; máx=16 anos).
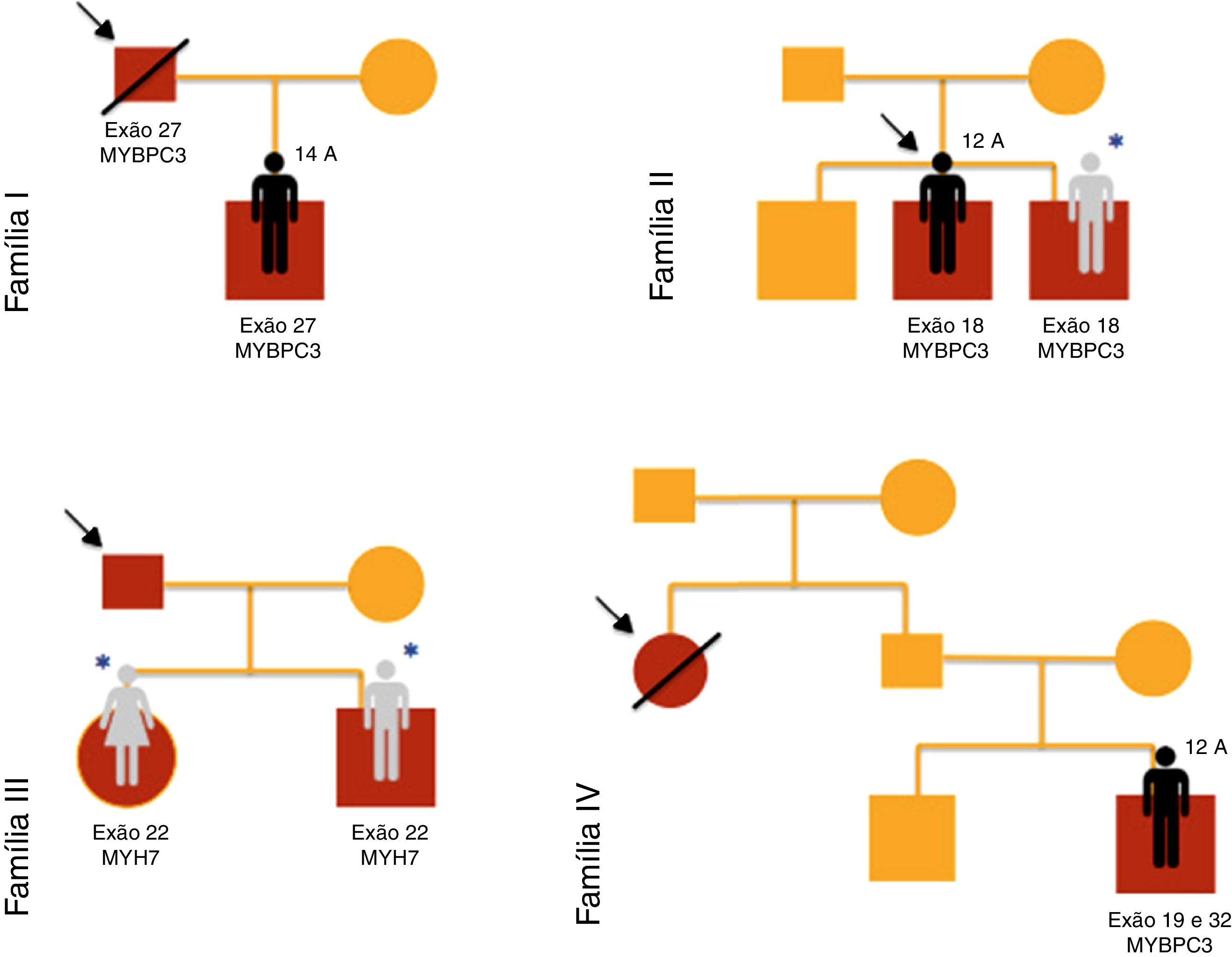
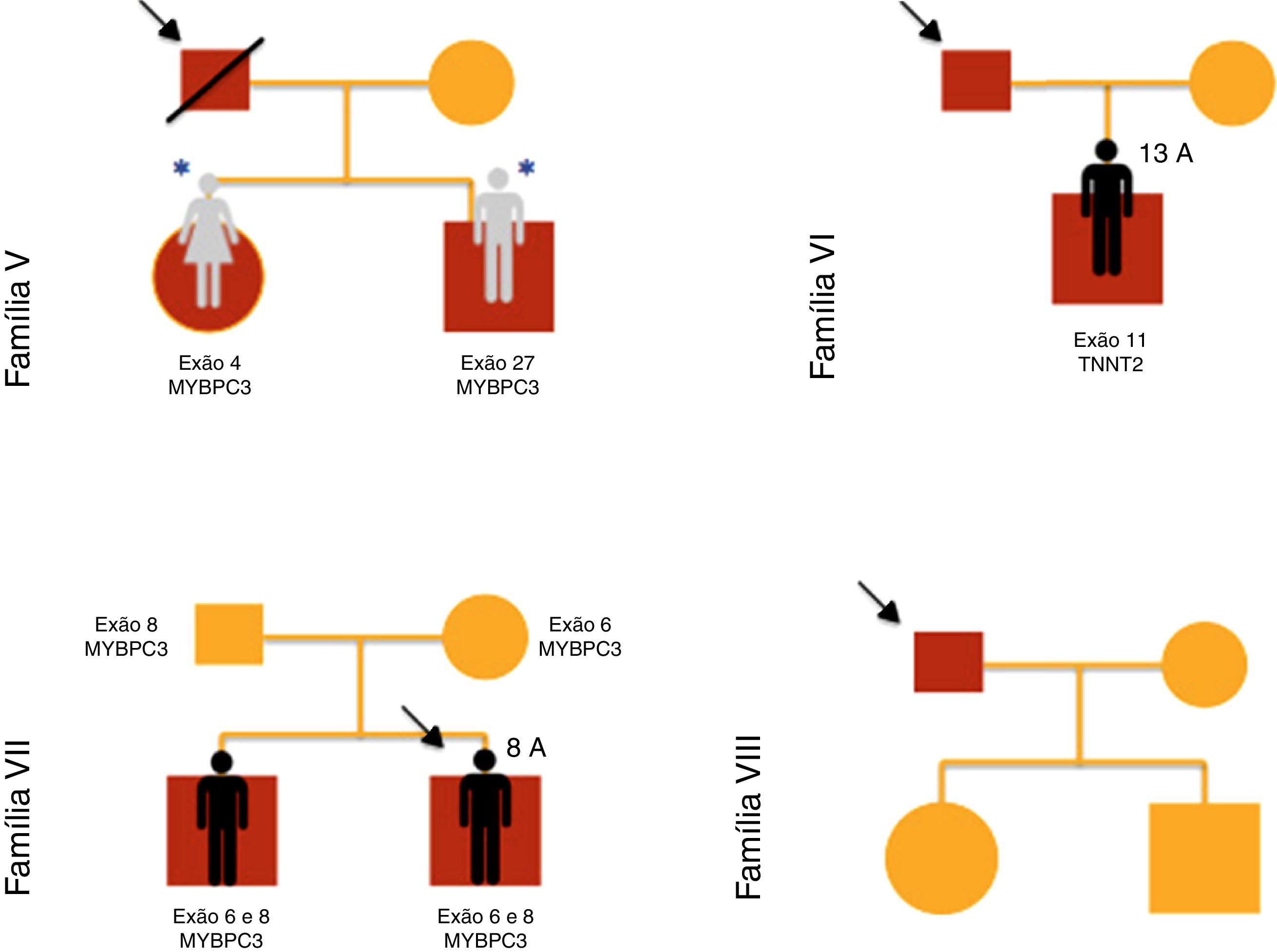
O motivo da referenciação dos três casos índex à consulta de cardiologia pediátrica foi, respetivamente: presença de alterações no ECG, toracalgia com o esforço e sopro cardíaco.
Nas famílias I, IV e V, registava‐se história familiar de morte súbita cardíaca.
Achados clínicos à inclusão (Tabela 1)À, data da primeira consulta, a maioria das crianças (n=16; 80%) estava assintomática. Em duas das crianças havia referência a um episódio de síncope inexplicada e outras duas referiam toracalgia com o esforço.
Características das crianças incluídas na amostra de MCH familiar
| Caso | Família | Caso índex | Género | Idade (anos) | Motivo referenciação | Sintomas | Síndrome malformativa | Fenótipo | Genótipo | Classificação |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | I | Pai | M | 14 | HF | Toracalgia com esforço | – | MCH | + | Familiar afetado |
| 2 | II | Irmão | M | 7 | HF | 0 | – | N | + | Familiar em risco |
| 3 | II | Próprio | M | 12 | Alterações do ECG | 0 | – | MCH | + | Caso índex |
| 4 | II | Irmão | M | 10 | HF | 0 | – | N | – | Familiar sem risco de doença |
| 5 | III | Pai | F | 5 | HF | 0 | – | N | + | Familiar em risco |
| 6 | III | Pai | M | 2 | HF | 0 | – | N | + | Familiar em risco |
| 7 | IV | Tia paterna | M | 11 | HF | 0 | – | N | – | Familiar sem risco de doença |
| 8 | IV | Tia paterna | M | 12 | HF | Síncope | – | MCH | + | Familiar afetado |
| 9 | V | Pai | F | 5 | HF | 0 | – | N | + | Familiar em risco |
| 10 | V | Pai | M | 7 | HF | 0 | – | N | + | Familiar em risco |
| 11 | VI | Pai | M | 13 | HF | Síncope | – | MCH | + | Familiar afetado |
| 12 | VII | Próprio | M | 8 | Toracalgia com esforço | Toracalgia com esforço | – | MCH | + | Caso índex |
| 13 | VII | Irmão | M | 10 | HF | 0 | – | N | + | Familiar em risco |
| 14 | VIII | Pai | F | 14 | HF | 0 | – | N | – | Familiar sem risco de doença |
| 15 | VIII | Pai | M | 7 | HF | 0 | – | N | – | Familiar sem risco de doença |
| 16 | IX | Irmão | F | 0,08 | HF | 0 | Síndrome LEOPARD | N | ? | Familiar em risco |
| 17 | IX | Irmão | M | 5 | HF | 0 | Síndrome LEOPARD | N | + | Familiar em risco |
| 18 | IX | Próprio | M | 0,1 | Sopro | 0 | Síndrome LEOPARD | MCH | + | Caso índex |
| 19 | X | Pai | M | 16 | HF | 0 | – | MCH | + | Familiar afetado |
| 20 | X | Pai | M | 11 | HF | 0 | – | N | – | Familiar sem risco de doença |
F: feminino; HF: história familiar; M: masculino; MCH: miocardiopatia hipertrófica; N: normal.
Sete (50%) dos 14 portadores de mutação apresentavam fenótipo positivo na primeira avaliação. A função sistólica do VE encontrava‐se conservada em todos eles.
Nenhum dos doentes apresentava obstrução sistémica significativa em repouso.
Num dos doentes elicitou‐se obstrução intraventricular esquerda significativa com o esforço, tendo sido medicado com bloqueador‐beta e aconselhado a restrição da atividade física.
Oito crianças (40%) eram portadoras de mutação de um gene sarcomérico, embora sem manifestação fenotípica de doença à apresentação. Foram consideradas em risco de desenvolvimento de MCH.
Cinco dos familiares avaliados (25%) apresentavam fenótipo e genótipo negativos, pelo que se considerou não haver risco de desenvolvimento de MCH, tendo sido dispensados de seguimento.
Estudo genético (Tabela 2)À, data da primeira consulta, 14 crianças (70%) eram portadoras de mutação de um ou mais genes sarcoméricos (80% sexo masculino; idade mediana=8 anos; min=1 mês; máx=16 anos).
Mutações de genes sarcoméricos identificadas nos doentes em estudo
| Caso | Família | Gene mutado | Alteração cDNA | Alteração proteína | Significado clínico | |
|---|---|---|---|---|---|---|
| 1 | I | Exão 27 do gene MYBPC3 | c.2864_2865delCT | p.Pro955ArgfsX95 | Mutação patogénica HGMD CD982813 | |
| 2 | II | Exão 18 do gene MYBPC3 | c.1684G>A | p.Ala562Thr | Variante genética de significado incerto | PolyPhen‐2: Possibly Damaging |
| Mutation Taster: Disease Causing | ||||||
| 3 | II | Exão 18 do gene MYBPC3 | c.1684G>A | p.Ala562Thr | Variante genética de significado incerto | PolyPhen‐2: Possibly Damaging |
| Mutation Taster: Disease Causing | ||||||
| 4 | II | – | – | – | ||
| 5 | III | Exão 22 do gene MYH7 | c.2539_2541delAAG | p.Lys847del | Mutação patogénica HGMD CD046025 HGMD CM0910620 | |
| 6 | III | Exão 22 do gene MYH7 | c.2539_2541delAAG | p.Lys847del | Mutação patogénica HGMD CD046025 HGMD CM0910620 | |
| 7 | IV | – | – | – | ||
| 8 | IV | Exão 19 do gene MYBPC3 | c.1828G>A e | p.Asp610Asn | Variante genética de significado incerto | PolyPhen‐2: Probably Damaging |
| Mutation Taster: Disease Causing | ||||||
| Exão 32 do gene MYBPC3 | c.3617G>A | p.Gly1206Asp | Mutação patogénica HGMD CM057198 | |||
| 9 | V | Exão 4 do gene MYBPC3 | c.458C>A | p.Pro153His | Mutação não descrita | PolyPhen‐2: Probably Damaging |
| Mutation Taster: Disease Causing | ||||||
| 10 | V | Exão 27 do gene MYBPC3 | c.2827C>T | p.Arg943STOP | Mutação patogénica HGMD CM032959 | |
| 11 | VI | Exão 11 do gene TNNT2 | c.458_489del3 | p.Glu163del | Mutação patogénica HGMD CD9518665 | |
| 12 | VII | Exão 6 do gene MYBPC3 | c.772G>A | p.Glu258Lys | Mutação patogénica HGMD CM981322 | |
| Exão 8 do gene MYBPC3 | c.836G>C | p.Gly279Ala | Variante genética de significado incerto HGMD CM031257 | PolyPhen‐2: Benign | ||
| Mutation Taster: Polymorphism | ||||||
| 13 | VII | Exão 6 do gene MYBPC3 | c.772G>A | p.Glu258Lys | Mutação patogénica HGMD CM981322 | |
| Exão 8 do gene MYBPC3 | c.836G>C | p.Gly279Ala | Variante genética de significado incerto HGMD CM031257 | PolyPhen‐2: Benign | ||
| Mutation Taster: Polymorphism | ||||||
| 14 | VIII | – | – | – | ||
| 15 | VIII | – | – | – | ||
| 16 | IX | Aguarda | Aguarda | Aguarda | ||
| 17 | IX | Exão 17 do gene MYBPC3 | c.1519G>A | p.Gly507Arg | Variante genética de significado incerto HGMD CM032598 | PolyPhen‐2: Probably Damaging |
| Mutation Taster: Disease Causing | ||||||
| 18 | IX | Exão 17 do gene MYBPC3 | c.1519G>A | p.Gly507Arg | Variante genética de significado incerto HGMD CM032598 | PolyPhen‐2: Probably Damaging |
| Mutation Taster: Disease Causing | ||||||
| 19 | X | Exão 4 do gene MYBPC3 | c.446C>A | p.Ala149Asp | Mutação não descrita | PolyPhen‐2: Benign |
| Mutation Taster: Polymorphism | ||||||
| Exão 2 do gene MYL3 | c.145G>T | p.Glu49STOP | Mutação não descrita | Mutation Taster: Disease causing | ||
| 20 | X | – | – | – | ||
MYBPC3: myosin binding protein C; MYH7: myosin heavy chain; MYL3: myosin light chain 3; TNNT2: cardiac troponinT.
Verificaram‐se mutações dos genes MYBPC3 (n=14; 78%), MYH7 (n=2; 11%), TNNT2 (n=1; 5,5%) e MYL3 (n=1; 5,5%).
Um dos doentes (caso 8, família IV) apresentava em heterozigotia duas mutações no gene MYBPC3 (exões 19 e 32), uma delas previamente descrita em doentes com MCH (p.Gly1206Asp) e a outra uma variante genética de significado incerto (p.Asp610Asn). No entanto, esta mutação foi predita como provavelmente patogénica pelos métodos PolyPhen‐2 e Mutation Taster.
Nos irmãos 12 e 13 (família VII) estavam presentes duas mutações no gene MYBPC3 (exão 6 e 8), uma delas previamente descrita em doentes com MCH (p.Glu258Lys) e a outra (p.Gly279Ala) predita como provavelmente benigna pelos métodos PolyPhen‐2 e Mutation Taster.
No caso 19 (família X) foram encontradas em heterozigotia a mutação p.Ala149Asp no exão 4 do gene MYBPC3 e a mutação p.Glu49STOP no exão 2 do gene MYL3. A mutação p.Ala149Asp não foi previamente descrita em doentes com MCH e foi predita como provavelmente benigna pelos métodos PolyPhen‐2 e Mutation Taster. A mutação p.Glu49STOP não foi previamente descrita em doentes com MCH, mas dada a consequência para a sequência proteica assumiu‐se que pudesse constituir a causa genética da patologia observada.
Na família IX, os casos 17 e 18 apresentavam estudo genético positivo para síndrome de LEOPARD, com identificação de mutação patogénica no exão12 do gene PTPN11 (c.1403C>T; p.Thr468Met). Foi‐lhes identificada em heterozigotia uma mutação no exão 17 do gene MYBPC3 (p.Gly507Arg), uma variante genética de significado incerto, predita como provavelmente patogénica pelos métodos PolyPhen‐2 e Mutation Taster.
Estratificação do risco de morte súbita (Tabela 3)Foi realizada análise do risco de morte súbita nos doentes com fenótipo e genótipo positivos. Três dos sete doentes não apresentavam qualquer fator de risco.
Estratificação do risco de morte súbita dos doentes em seguimento
| Caso | Família | Fator de risco «clássico» | Genótipo | Obstrução TSVE (Δmáx TSVE) | Score de risco ECG |
|---|---|---|---|---|---|
| 1 | I | AF morte súbita | – | – | 8 |
| 3 | II | SIVd>30mm | – | – | 8 |
| 8 | IV | Síncope; AF morte súbita | Genótipo complexo* | – | 7 |
| 11 | VI | Síncope; SIVd>30mm; AF morte súbita | – | – | 12 |
| 12 | VII | – | – | SAM, Δintraventricular=100mmHg (esforço) | 3 |
| 18 | IX | – | – | – | 4 |
| 19 | X | – | – | – | 3 |
AF: antecedentes familiares; ECG: eletrocardiograma; SAM: Systolic anterior movement; SIVd: dimensão diastólica do septo interventricular; TSVE: trato de saída do ventrículo esquerdo.
Num dos doentes verificava‐se um genótipo complexo, isto é, coexistência de mais do que uma mutação de um gene sarcomérico.
Quatro dos doentes apresentavam um score de risco eletrocardiográfico≥6 (Tabela 4). Dois dos três doentes com score de risco eletrocardiográfico<6 não apresentavam outros fatores de risco para morte súbita.
Score de risco eletrocardiográfico
| Caso 1 | Caso 3 | Caso 8 | Caso 11 | Caso 12 | Caso 18 | Caso 19 | ||
|---|---|---|---|---|---|---|---|---|
| Desvio eixo QRS | 1 ponto | 1 | 0 | 0 | 1 | 0 | 1 | 0 |
| Inversão T nas derivações membros* | 1 ponto | 0 | 1 | 1 | 1 | 0 | 0 | 0 |
| Inversão T nas derivações precordiais* | 2 pontos | 2 | 1 | 1 | 1 | 0 | 0 | 0 |
| Depressão ST>2mm | 2 pontos | 0 | 0 | 1 | 2 | 0 | 0 | 0 |
| S dominante V4 | 2 pontos | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| >7,7 mV=1 ponto | 0 | 3 | 1 | 3 | 0 | 0 | 0 | |
| Soma da amplitude QRS derivações dos membros | >10mV=2 pontos | |||||||
| >12mV=3 pontos | ||||||||
| Produto amplitude‐duração QRS nas 12 derivações | >2,2mV.s=1 ponto | 3 | 3 | 3 | 3 | 3 | 3 | 3 |
| >2,5mV.s=2 pontos | ||||||||
| >3mV.s=3 pontos | ||||||||
| QTc>440ms | 1 ponto | 1 | 0 | 0 | 1 | 0 | 0 | 0 |
| Score de risco | 8 | 8 | 7 | 12 | 3 | 4 | 3 |
O tempo médio de seguimento destes doentes foi de 3,5±0,8 anos (min=6 meses; máx=9,5 anos).
Características dos doentes à data da última consulta
| Caso | Família | Idade (anos) | Fenótipo | Sintomas | Complicações | Classificação |
|---|---|---|---|---|---|---|
| 1 | I | 16 | MCH | 0 | – | Familiar afetado |
| 2 | II | 10 | MCH | 0 | – | Familiar afetado |
| 3 | II | 19 | MCH | 0 | – | Caso índex |
| 5 | III | 6 | N | Palpitações | – | Familiar em risco |
| 6 | III | 4 | N | 0 | – | Familiar em risco |
| 8 | IV | 14 | MCH | 0 | – | Familiar afetado |
| 9 | V | 8 | N | 0 | – | Familiar em risco |
| 10 | V | 9 | N | 0 | – | Familiar em risco |
| 11 | VI | 17 | MCH | 0 | CDI | Familiar afetado |
| 12 | VII | 18 | MCH | 0 | – | Caso índex |
| 13 | VII | 15 | MCH | 0 | – | Familiar afetado |
| 16 | IX | 3 | N | 0 | – | Familiar em risco |
| 17 | IX | 12 | N | 0 | – | Familiar em risco |
| 18 | IX | 7 | MCH | 0 | – | Caso índex |
| 19 | X | 17 | MCH | 0 | – | Familiar afetado |
CDI: cardiodisfibrilhador implantável; F: feminino; M: masculino; MCH: miocardiopatia hipertrófica; N: fenótipo normal; NYHA: New York Heart Association.
Durante este período, duas das crianças fenótipo negativo portadoras de mutação (gene MYBPC3) desenvolveram MCH, aos 10 e 15 anos de idade (28% de taxa de penetrância).
Todos os doentes diagnosticados com MCH à primeira avaliação mantinham o diagnóstico à data da última consulta.
Não se verificaram óbitos durante o período de seguimento. Um dos doentes foi submetido a implantação de cardioversor‐desfibrilhador como prevenção primária, após um episódio de taquicardia ventricular não mantida registado em Holter de 24 horas.
Este doente apresentava três fatores de risco clássicos (síncope, hipertrofia ventricular esquerda extrema e história de morte súbita de tia paterna com o diagnóstico de MCH) e um score de risco eletrocardiográfico=12 (Tabela 3).
DiscussãoA MCH tem uma incidência anual estimada de 0,3‐0,5 por cada 100000 crianças3,11.
Na população adulta a MCH familiar resulta, em 60% dos casos, de mutações nos genes das proteínas sarcoméricas3. Em pediatria, no entanto, esta patologia tem uma etiologia mais complexa e heterogénea.
No Pediatric Cardiomyopathy Registry, numa população de 855 doentes com MCH, a etiologia era conhecida em apenas 25,8%: em 9% dos casos registava‐se a associação a síndromes malformativos, em 8,7% a doenças do metabolismo e em 7,5% a doenças neuromusculares12. Estima‐se que em cerca de um terço dos casos de MCH pediátrica a etiologia seja familiar, resultante de mutações dos genes que codificam as proteínas sarcoméricas9.
Estudo genético na cardiomiopatia hipertrófica familiarForam já descritas mais de 1400 mutações nos 11 genes que codificam as proteínas dos miofilamentos sarcoméricos e do disco‐Z5.
Em cerca de 50‐60% dos casos de MCH familiar é possível identificar uma mutação sarcomérica5. Quando uma mutação patogénica é identificada numa família, o diagnóstico genético é uma medida de rastreio familiar custo‐efetiva e eficaz.
O objetivo deste último é o de identificar a ocorrência ou o risco de desenvolvimento da doença nos familiares em primeiro grau do caso índex, para início precoce de terapêutica e estratificação do risco de morte súbita cardíaca.
As mais recentes normas de orientação da ESC e da ACCF/AHA recomendam a avaliação dos familiares em idade pediátrica a partir dos 10‐12 anos3,4. No entanto, em famílias com patologia de apresentação precoce poderá ser apropriada a avaliação clínica e genética anterior a esta idade3.
Atualmente, o valor do teste genético assenta sobretudo na decisão de manter ou dispensar do seguimento clínico e ecocardiográfico os familiares do caso índex3,4.
Na nossa amostra, cinco (25%) das crianças avaliadas apresentavam fenótipo e genótipo negativos, e foram dispensadas de seguimento regular. Esta informação tranquiliza os familiares não afetados quanto à possibilidade de desenvolvimento de doença e liberta os serviços de consultas desnecessárias.
Por outro lado, as crianças com genótipo positivo foram divididas em dois grupos: aquelas com fenótipo positivo (n=7; 35%) – familiares afetados – e aquelas com fenótipo negativo (n=8; 40%) – familiares em risco.
Para classificação das variantes de ADN quanto à sua patogenicidade, foram consultadas as bases de dados da Clinvar e da HGMD.
Em oito dos casos, as mutações encontradas tinham sido previamente descritas como patogénicas.
Nas situações em que a mutação não tinha sido previamente descrita (n=3) e para as variantes genéticas de significado incerto (n=7), foram utilizados os métodos PolyPhen‐2 e Mutation Taster, para aferição da sua relevância funcional.
Optámos por incluir neste estudo os doentes pertencentes à família IX que, apesar de terem o diagnóstico genético de síndrome de LEOPARD e esta ser por si só explicativa do fenótipo de MCH, apresentavam paralelamente em heterozigotia uma mutação no exão 17 do gene MYBPC3 (p.Gly507Arg). Esta constitui‐se como uma variante genética de significado incerto, predita como provavelmente patogénica pelos métodos PolyPhen‐2 e Mutation Taster. Neste contexto, a patogenicidade da mutação sarcomérica encontrada permanece por esclarecer.
Foi nossa política manter em seguimento os doentes com variantes genéticas de significado incerto e aqueles apresentando mutações não anteriormente descritas como estando associadas à ocorrência de MCH, mesmo na ausência de manifestações fenotípicas de doença («familiares em risco»).
No entanto, para além da avaliação sequencial, o manuseio clínico dos portadores de mutação de genes sarcoméricos sem fenótipo de doença não está estabelecido3.
De acordo com as mais recentes recomendações internacionais, as crianças genótipo positivo/fenótipo negativo devem ser avaliadas cada 12‐18 meses, enquanto os adultos apenas a cada 2‐5 anos3.
Em doentes portadores de mutação de um gene sarcomérico mas sem expressão fenotípica de doença, o risco de eventos cardíacos adversos é baixo, tendo sido reportada num estudo recente uma taxa de morte súbita cardíaca de 0,13% por pessoa‐ano nesta população13.
Morte súbita e estratificação do risco de morte súbita cardíacaEstima‐se que o risco anual de morte súbita cardíaca em doentes com MCH ronde 1%13. No entanto, um estudo dinamarquês, realizado entre 2000‐2006, revelou um risco de morte súbita cardíaca em contexto de MCH<0,1% por pessoa‐ano, em doentes com idades compreendidas entre 1‐35 anos14.
As mais recentes recomendações da ESC postulam como fatores de risco major para a ocorrência de morte súbita em crianças com MCH a hipertrofia ventricular esquerda extrema (definida como uma espessura ventricular esquerda máxima≥30mm ou Z‐score≥6), ocorrência de síncope inexplicada, taquicardia ventricular não mantida e história familiar de morte súbita cardíaca3.
Na nossa amostra, optou‐se pela estratificação do risco de morte súbita apenas nos familiares afetados, tal como preconizado nas atuais recomendações4.
Foram considerados, a par dos fatores de risco clássicos, outros fatores de risco descritos na literatura: presença de obstrução no TSVE9 e genótipo complexo (coexistência de mais do que uma mutação de um gene sarcomérico)3,15.
Foi ainda aplicado o score de risco eletrocardiográfico descrito por Östman‐Smith et al., que num estudo desenvolvido em adultos com MCH se associou significativamente à ocorrência morte súbita na população estudada, com elevadas sensibilidade (85%) e especificidade (100%)9.
Este modelo preditivo avalia a presença de desvio do eixo QRS, inversão patológica das ondas T, depressão do segmento ST, S dominante em V4, soma da amplitude de QRS nas derivações dos membros, produto amplitude‐duração de QRS nas 12 derivações e QTc.
É interessante notar que, na nossa amostra, o único doente com complicações (implantação de CDI) apresentava, além de três fatores de risco clássicos, o score de risco eletrocardiográfico mais elevado da população em estudo.
Expressão fenotípica da miocardiopatia hipertróficaA expressão clínica da MCH é determinada por uma complexa interação de fatores genéticos, epigenéticos e ambientais. Tal como outras patologias de transmissão autossómica dominante, demonstra marcada variabilidade fenotípica, mesmo no seio das famílias afetadas.
Foram propostas como manifestações pré‐fenotípicas detetáveis em portadores de mutações sarcoméricas a presença de criptas miocárdicas, o alongamento dos folhetos mitrais, a disfunção diastólica, o aumento da deposição de colagénio e a presença de fibrose miocárdica3,4.
A heterogeneidade clínica desta patologia é também longitudinal: a penetrância da expressão fenotípica aumenta com a idade, mantendo‐se no entanto inferior a 100%5.
Um estudo recente descreveu uma incidência de MCH manifesta em portadores de mutações sarcoméricas com idade inferior a 40 anos<0,10% por pessoa‐ano13.
Diversos estudos pediátricos demonstraram uma penetrância da expressão fenótipica que varia entre 6‐31% em crianças portadoras de mutações de genes sarcoméricos, após um período de seguimento máximo de 12 anos2,16.
Na nossa amostra, duas das crianças fenótipo negativo portadoras de mutação desenvolveram o fenótipo de MCH após 3,5±0,8 anos de seguimento, aos dez e 15 anos. A taxa de penetrância resultante (28%) enquadra‐se nas anteriormente descritas na literatura.
Este facto sublinha a importância do seguimento longitudinal dos doentes portadores de mutações dos genes sarcoméricos, e perspetiva o desenvolvimento futuro de terapêuticas modeladoras da expressão da doença.
Limitações do estudoAs limitações deste estudo são inerentes ao seu desenho retrospetivo e ao reduzido tamanho da amostra.
É importante ressaltar que a taxa de conversão fenotípica depende provavelmente de uma multiplicidade de fatores, entre os quais o sexo, a raça e o genótipo. Na nossa amostra estão representadas apenas crianças de raça caucasiana e com mutações em quatro dos genes sarcoméricos (MYBPC3, MYH7, TNNT2 e MYL3).
O reduzido número de complicações ocorridas na nossa amostra não nos permitiu estabelecer associações entre estas e os fatores de risco identificados.
ConclusãoQuando uma mutação patogénica é identificada numa família, o teste genético é uma medida eficaz de rastreio familiar. Este último permite aferir quais os familiares em risco de desenvolvimento de doença e aqueles que podem ser dispensados do seguimento.
Na nossa amostra, a penetrância de MCH em crianças com genótipo positivo e fenótipo normal à apresentação foi de 28% após 3,5 anos de seguimento. Este facto sublinha a importância da avaliação longitudinal dos portadores de mutação de genes sarcoméricos, independentemente da presença de fenótipo patológico.
Responsabilidades éticasProteção de pessoas e animaisOs autores declaram que para esta investigação não se realizaram experiências em seres humanos e/ou animais.
Confidencialidade dos dadosOs autores declaram ter seguido os protocolos do seu centro de trabalho acerca da publicação de dados de pacientes.
Direito à privacidade e consentimento escritoOs autores declaram que não aparecem dados que permitam a identificação de pacientes neste artigo.
Conflito de interessesOs autores declaram não haver conflito de interesses.
