







Hypertrophic cardiomyopathy (HCM) is most often of autosomal dominant inheritance with incomplete penetrance and variable expression. The main purpose of family screening is to identify relatives with unrecognized HCM and to monitor those at risk for disease, in order to minimize complications and to assess risk of sudden cardiac death. The ESC and ACCF/AHA guidelines on the diagnosis and management of HCM recommend the screening of child relatives from the age of 10-12 years.
ObjectivesWe studied the outcome of clinical screening and genetic testing of child probands and relatives (<18 years of age) from families with HCM and assessed the age-related penetrance of HCM during the follow-up of these young relatives.
Methods and ResultsTwenty patients from ten families were included between 2004 and 2013, consisting of three probands and 17 first-degree relatives (80% male; median age 10 years). Fourteen child relatives were mutation carriers (70%; median age eight years). Seven (50%) of the 14 mutation carriers were diagnosed with HCM at initial assessment. At-risk child relatives were defined as those with a positive mutation but a negative phenotype at enrollment.
After 3.5±0.8 years of follow-up, two of the phenotype-negative mutation carriers developed HCM at 10 and 15 years of age (28% penetrance rate).
ConclusionsThe penetrance of HCM in phenotype-negative child relatives was 28% after 3.5 years of follow-up. This underlines the need for long-term monitoring of mutation carriers irrespective of the presence of a positive phenotype.
A miocardiopatia hipertrófica (MCH) é uma patologia com transmissão essencialmente autossómica dominante, expressão clínica variável e penetrância incompleta. O rastreio familiar tem por objetivo identificar a ocorrência ou o risco de desenvolvimento da doença nos parentes em primeiro grau do caso índex. As normas de orientação da ESC e da ACCF/AHA recomendam a avaliação dos familiares em idade pediátrica a partir dos 10-12 anos.
ObjetivosAvaliaram-se os resultados de um programa de rastreio pediátrico de MCH familiar e o valor preditivo do seu estudo genético. Foi ainda aferida a penetrância fenotípica ao longo do tempo de seguimento destas crianças.
Métodos e resultadosForam incluídas 20 pertencentes a dez famílias (2004-2013). Três das crianças constituíram-se como o caso índex, sendo as restantes parentes em primeiro grau de um doente com MCH (80% sexo masculino; idade mediana=10 anos). Catorze crianças eram portadoras de mutação de um gene sarcomérico (70%; idade mediana=8 anos). Sete (50%) dos 14 portadores de mutação apresentavam fenótipo positivo na primeira avaliação.
Foram definidos como «familiares em risco» aqueles com teste genético positivo, mas com fenótipo normal à apresentação. Após 3,5±0,8 anos de seguimento, duas das crianças fenótipo negativo portadoras de mutação (gene MYBPC3) desenvolveram MCH, aos dez e 15 anos de idade (28% de taxa de penetrância).
ConclusõesA penetrância de MCH em crianças com fenótipo normal à apresentação foi de 28% após 3,5 anos de seguimento. Tal sublinha a importância da avaliação longitudinal dos portadores de mutação de genes sarcoméricos, independentemente da presença de fenótipo patológico.
American College of Cardiology Foundation/American Heart Association
body mass index
body surface area
confidence interval
electrocardiogram
European Society of Cardiology
implantable cardioverter-defibrillator
hypertrophic cardiomyopathy
Human Gene Mutation Database
left ventricular
left ventricular outflow tract
polymerase chain reaction
corrected QT
relative risk
systolic anterior movement
sudden cardiac death
ventricular tachycardia
Hypertrophic cardiomyopathy (HCM) is most often of autosomal dominant inheritance with variable expression and age-related incomplete penetrance.1
Its clinical expression is heterogeneous, ranging from asymptomatic to severe heart failure symptoms or sudden cardiac death (SCD).2
The main purpose of family screening is to identify first-degree relatives of the proband with or at risk of developing the disease.
The latest guidelines of the European Society of Cardiology (ESC) and of the American College of Cardiology Foundation/American Heart Association (ACCF/AHA) recommend screening of child relatives from the age of 10-12 years.3,4
It is estimated that a mutation in the genes coding for sarcomeric proteins can be identified in 50-60% of cases of familial HCM.5 However, in children with a negative phenotype, the prognostic value of identifying such mutations is unclear.
We studied the outcome of screening for familial HCM in a tertiary pediatric cardiology reference center and assessed the predictive value of genetic testing. We also analyzed the age-related penetrance of the disease during the follow-up of these young relatives.
MethodsStudy populationWe analyzed all cases of familial HCM followed in specialist consultations in a tertiary pediatric cardiology reference center between 2004 and 2013. All child relatives under the age of 18 of a proband with a positive genetic test for sarcomeric gene mutations were included.
Referral of first-degree relatives of a proband diagnosed with HCM was made mainly following cardiology consultation in the same center; when the proband was a child, siblings were referred for pediatric cardiology consultation and the parents for cardiology consultation.
All patients were also referred for genetic consultation in the same center.
Clinical assessment and genetic testingInitial assessment of the study population included clinical observation, 12-lead resting ECG, transthoracic echocardiogram and genetic screening for the eight most common sarcomeric gene mutations associated with HCM (in MYH7, MYL2, MYL3, MYBPC3, TNNI3, TNNT2, TPM1 and ACTC1).
Screening for mutations in the above genes (entire coding region, including intron/exon boundaries) was performed using polymerase chain reaction (PCR) technology with direct sequencing (combination of next-generation sequencing with a minimum of 30× coverage and Sanger sequencing) of the PCR products. This method has an analytical sensitivity of 99% for the detection of nucleotide substitutions and small deletions and insertions.
The ClinVar database and the Human Gene Mutation Database (HGMD) were used to classify the pathogenicity of DNA variants. The bioinformatics tools PolyPhen-2 and Mutation Taster were used to predict the disease-causing potential of mutations that had not been previously described or genetic variants of uncertain significance by assessing their functional effects.
Children with more than one mutation were classified as having a compound genotype.
Two-dimensional, M-mode and Doppler echocardiography were performed in accordance with the guidelines of the American Society of Echocardiography.6
Dimensions of the cardiac chambers, interventricular septum and left ventricular (LV) posterior wall, mitral valve systolic anterior movement (SAM) and LV outflow tract (LVOT) gradient were determined, at rest and during the Valsalva maneuver. LVOT obstruction was defined as a resting gradient of ≥30 mmHg.3
An echocardiographic diagnosis of HCM was made when the maximum LV posterior wall thickness was greater than twice the standard deviation of the predicted mean adjusted for body surface area (BSA).3 BSA was calculated according to the Haycock formula.7
The electrocardiogram (ECG) was analyzed for QRS axis deviation, T-wave inversion of >1 mm, ST-segment depression of >2 mm and S wave > R wave in V4. Overall QRS amplitude, limb-lead QRS amplitude sum, 12-lead QRS amplitude-duration product, and corrected QT (QTc) according to Bazett's formula were calculated.8
SCD risk was stratified using the model proposed by Östman-Smith et al.9 The patients were scored from 1 to 3 on the eight parameters analyzed (maximum score 14), a total score of ≥6 indicating high risk. Although this predictive model was developed for adults with HCM, the authors used the same model in a pediatric population and reported similar predictive value (unpublished study, presented at the 46th Annual Meeting of the Association for European Paediatric and Congenital Cardiology, Istanbul, Turkey, 23-26 May 2012).
The following risk factors for SCD were also analyzed10:
- •
family history of SCD: non-traumatic premature death (at age <40 years); death within an hour of symptom onset in the absence of previous symptoms, including unexpected or unwitnessed nocturnal death or equivalent, such as need for cardiopulmonary resuscitation or appropriate implantable cardioverter-defibrillator (ICD) shock;
- •
unexplained syncope of non-neurocardiogenic etiology;
- •
nonsustained ventricular tachycardia (VT): one or more episodes of ≥3 consecutive ventricular extrasystoles with heart rate of >120 bpm, lasting <30 s during exercise testing or 24-h Holter monitoring;
- •
severe LV hypertrophy: maximum LV wall thickness of ≥30 mm or z-score of ≥6.3.
Individuals with positive genotype and phenotype were classified as affected and followed in pediatric cardiology consultations every six months.
Carriers of sarcomeric gene mutations, genetic variants of uncertain significance or mutations not previously described as associated with HCM and with no phenotypic manifestations of the disease were classified as at risk of developing HCM. Genetic study was pending in one case, who was considered at risk of developing the disease. These children were followed in annual consultations.
Those with a negative phenotype and no mutation were not considered at risk of HCM and were discharged from follow-up.
Follow-up consultations included clinical assessment, 12-lead ECG and transthoracic echocardiogram, and 24-h Holter monitoring was requested whenever deemed clinically necessary.
All children aged >7 years underwent conventional exercise testing, and two also underwent exercise echocardiography.
Statistical analysisContinuous variables with normal distribution are presented as means and standard deviation and as medians, minimum and maximum otherwise. Categorical variables are expressed as frequencies and percentages.
ResultsTwenty children from ten families were included in this study of familial HCM (Figures 1–3), of whom three were probands and the remainder first-degree relatives of a patient with HCM (80% male; median age 10 years [1 month - 16 years]).
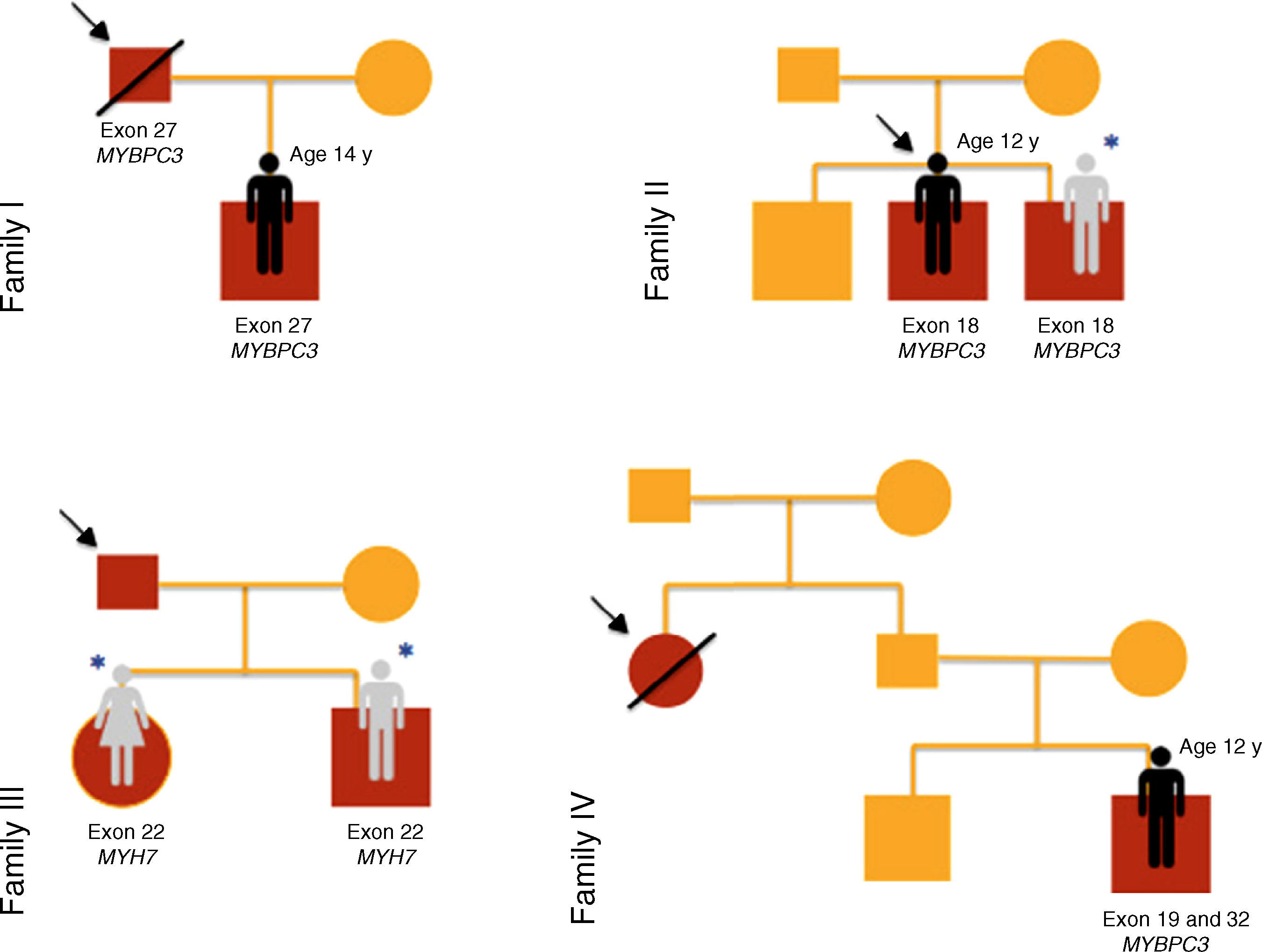
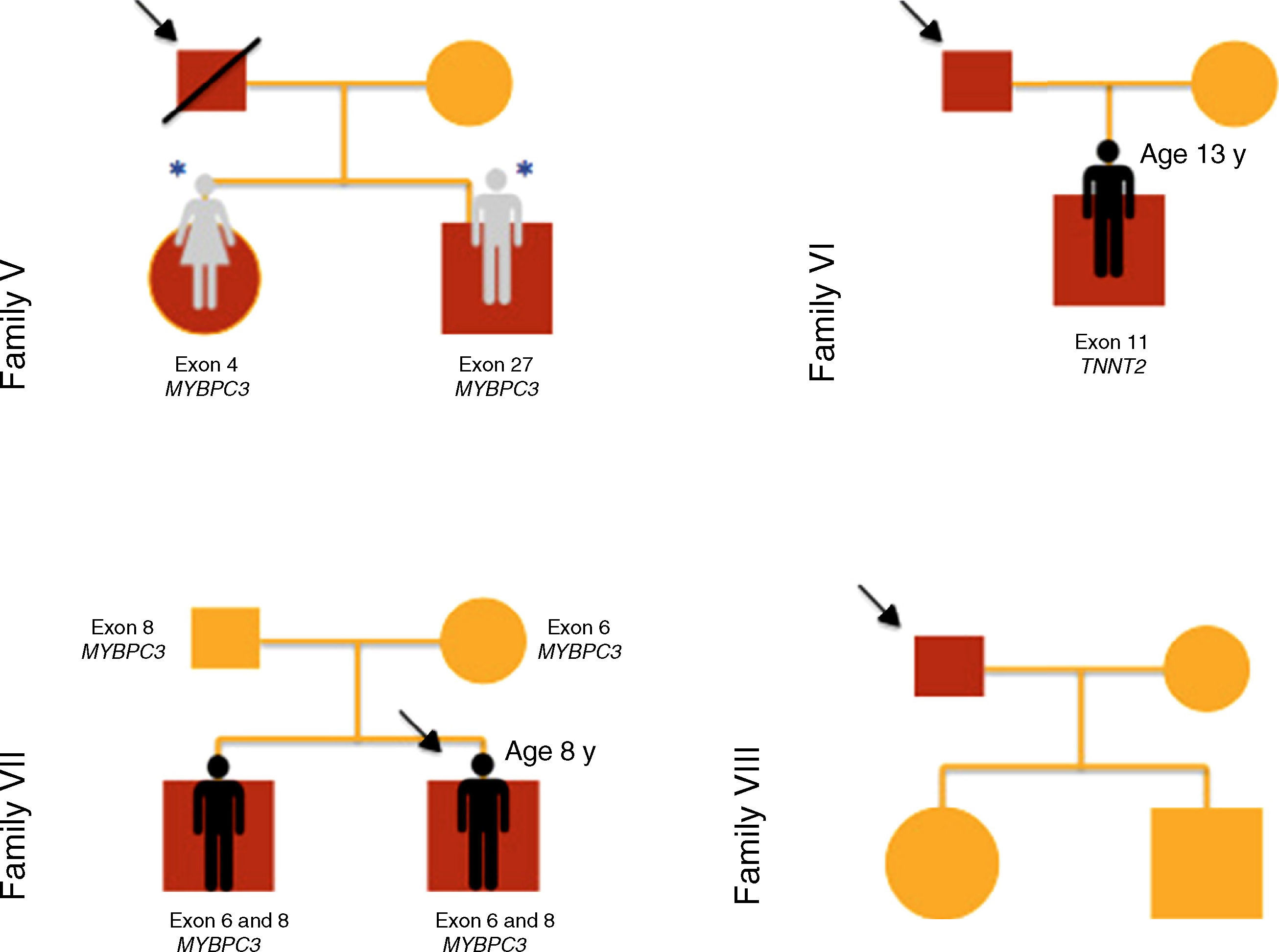
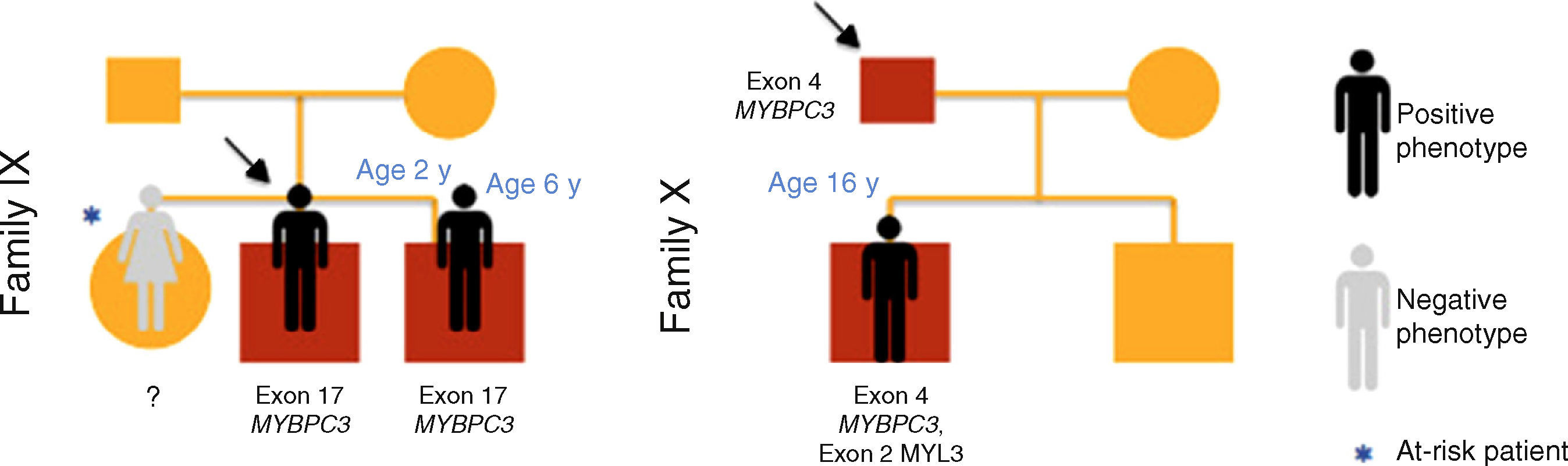
The reasons for referral for pediatric cardiology consultation of the three probands were ECG alterations, chest pain on exertion and heart murmur. There was a family history of SCD in families I, IV and V.
Clinical findings at recruitment (Table 1)Most children (n=16; 80%) were asymptomatic at initial assessment; an episode of unexplained syncope was reported in two children and two others reported chest pain on exertion.
Characteristics of the study population.
| Case | Family | Proband | Gender | Age (years) | Reason for referral | Symptoms | Malformation syndrome | Phenotype | Genotype | Classification |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | I | Father | M | 14 | FH | Chest pain on exertion | - | HCM | + | Affected |
| 2 | II | Brother | M | 7 | FH | 0 | - | N | + | At risk |
| 3 | II | Proband | M | 12 | ECG abnormalities | 0 | - | HCM | + | Proband |
| 4 | II | Brother | M | 10 | FH | 0 | - | N | - | Not at risk |
| 5 | III | Father | F | 5 | FH | 0 | - | N | + | At risk |
| 6 | III | Father | M | 2 | FH | 0 | - | N | + | At risk |
| 7 | IV | Paternal aunt | M | 11 | FH | 0 | - | N | - | Not at risk |
| 8 | IV | Paternal aunt | M | 12 | FH | Syncope | - | HCM | + | Affected |
| 9 | V | Father | F | 5 | FH | 0 | - | N | + | At risk |
| 10 | V | Father | M | 7 | FH | 0 | - | N | + | At risk |
| 11 | VI | Father | M | 13 | FH | Syncope | - | HCM | + | Affected |
| 12 | VII | Proband | M | 8 | Chest pain on exertion | Chest pain on exertion | - | HCM | + | Proband |
| 13 | VII | Brother | M | 10 | FH | 0 | - | N | + | At risk |
| 14 | VIII | Father | F | 14 | FH | 0 | - | N | - | Not at risk |
| 15 | VIII | Father | M | 7 | FH | 0 | - | N | - | Not at risk |
| 16 | IX | Brother | F | 0.08 | FH | 0 | LEOPARD syndrome | N | ? | At risk |
| 17 | IX | Brother | M | 5 | FH | 0 | LEOPARD syndrome | N | + | At risk |
| 18 | IX | Proband | M | 0.1 | Murmur | 0 | LEOPARD syndrome | HCM | + | Proband |
| 19 | X | Father | M | 16 | FH | 0 | - | HCM | + | Affected |
| 20 | X | Father | M | 11 | FH | 0 | - | N | - | Not at risk |
F: female; FH: family history; HCM: hypertrophic cardiomyopathy; M: male; N: normal.
Seven (50%) of the 14 mutation carriers presented a positive phenotype on initial assessment; LV systolic function was preserved in all of them.
No patient presented significant obstruction at rest. Significant LV obstruction was induced by exercise in one patient, who was medicated with beta-blockers and advised to restrict physical activity.
Eight children (40%) were carriers of a sarcomeric gene mutation, but had no phenotypic manifestation of the disease at initial assessment. They were considered at risk of developing HCM.
Five (25%) of the relatives assessed presented negative phenotype and genotype, and were thus considered not at risk of developing HCM and discharged from follow-up.
Results of genetic testing (Table 2)At initial assessment, 14 children (70%) - 80% male, median age eight years (one month - 16 years) - were carriers of one or more mutations in sarcomeric genes: MYBPC3 (n=14, 78%), MYH7 (n=2, 11%), TNNT2 (n=1, 5.5%) and MYL3 (n=1, 5.5%).
Sarcomeric gene mutations identified in the study population.
| Case | Family | Mutated gene | cDNA alteration | Protein alteration | Clinical significance | |
|---|---|---|---|---|---|---|
| 1 | I | Exon 27 of MYBPC3 | c.2864_2865delCT | p.Pro955ArgfsX95 | Disease-causing mutation. HGMD CD982813 | |
| 2 | II | Exon 18 of MYBPC3 | c.1684G>A | p.Ala562Thr | Genetic variant of uncertain significance | PolyPhen-2: possibly damaging |
| Mutation Taster: disease-causing | ||||||
| 3 | II | Exon 18 of MYBPC3 | c.1684G>A | p.Ala562Thr | Genetic variant of uncertain significance | PolyPhen-2: possibly damaging |
| Mutation Taster: disease-causing | ||||||
| 4 | II | - | - | - | ||
| 5 | III | Exon 22 of MYH7 | c.2539_2541delAAG | p.Lys847del | Pathogenic mutation HGMD CD046025 HGMD CM0910620 | |
| 6 | III | Exon 22 of MYH7 | c.2539_2541delAAG | p.Lys847del | Pathogenic mutation HGMD CD046025 HGMD CM0910620 | |
| 7 | IV | - | - | - | ||
| 8 | IV | Exon 19 of MYBPC3 | c.1828G>A e | p.Asp610Asn | Genetic variant of uncertain significance | PolyPhen-2: probably damaging |
| Mutation Taster: disease-causing | ||||||
| Exon 32 of MYBPC3 | c.3617G>A | p.Gly1206Asp | Pathogenic mutation HGMD CM057198 | |||
| 9 | V | Exon 4 of MYBPC3 | c.458C>A | p.Pro153His | Undescribed mutation | PolyPhen-2: Probably damaging |
| Mutation Taster: Disease-causing | ||||||
| 10 | V | Exon 27 of MYBPC3 | c.2827C>T | p.Arg943STOP | Pathogenic mutation HGMD CM032959 | |
| 11 | VI | Exon 11 of TNNT2 | c.458_489del3 | p.Glu163del | Pathogenic mutation HGMD CD9518665 | |
| 12 | VII | Exon 6 of MYBPC3 | c.772G>A | p.Glu258Lys | Pathogenic mutation HGMD CM981322 | |
| Exon 8 of MYBPC3 | c.836G>C | p.Gly279Ala | Genetic variant of uncertain significance HGMD CM031257 | PolyPhen-2: benign | ||
| Mutation Taster: polymorphism | ||||||
| 13 | VII | Exon 6 of MYBPC3 | c.772G>A | p.Glu258Lys | Pathogenic mutation HGMD CM981322 | |
| Exon 8 of MYBPC3 | c.836G>C | p.Gly279Ala | Genetic variant of uncertain significance HGMD CM031257 | PolyPhen-2: benign | ||
| Mutation Taster: polymorphism | ||||||
| 14 | VIII | - | - | - | ||
| 15 | VIII | - | - | - | ||
| 16 | IX | Pending | Pending | Pending | ||
| 17 | IX | Exon 17 of MYBPC3 | c.1519G>A | p.Gly507Arg | Genetic variant of uncertain significance HGMD CM032598 | PolyPhen-2: probably damaging |
| Mutation Taster: disease-causing | ||||||
| 18 | IX | Exon 17 of MYBPC3 | c.1519G>A | p.Gly507Arg | Genetic variant of uncertain significance HGMD CM032598 | PolyPhen-2: probably damaging |
| Mutation Taster: disease-causing | ||||||
| 19 | X | Exon 4 of MYBPC3 | c.446C>A | p.Ala149Asp | Undescribed mutation | PolyPhen-2: benign |
| Mutation Taster: polymorphism | ||||||
| Exon 2 of MYL3 | c.145G>T | p.Glu49STOP | Undescribed mutation | Mutation Taster: disease-causing | ||
| 20 | X | - | - | - | ||
MYBPC3: myosin binding protein C; MYL3: myosin light chain 3; MYH7: myosin heavy chain 7; TNNT2: cardiac troponin T.
One patient (case 8, family IV) presented two heterozygous mutations in MYBPC3 (exons 19 and 32), one previously described in HCM (p.Gly1206Asp) and the other a genetic variant of uncertain significance (p.Asp610Asn) but predicted to be pathogenic by PolyPhen-2 and Mutation Taster.
Two brothers (cases 12 and 13, family VII) presented two mutations in MYBPC3 (exon 6 and exon 8), one previously described in HCM (p.Glu258Lys) and the other (p.Gly279Ala) predicted to be benign by PolyPhen-2 and Mutation Taster.
In case 19 (family X) a heterozygous mutation (p.Ala149Asp) in exon 4 of MYBPC3 and another mutation (p.Glu49STOP) in exon 2 of MYL3 were detected. The mutation p.Ala149Asp has not been previously described in HCM but is predicted to be benign by PolyPhen-2 and Mutation Taster. The mutation p.Glu49STOP has also not been described in HCM, but given the consequences for the protein sequence, it is assumed that it could be disease-causing.
Cases 17 and 18 (family IX) presented positive genetic study for LEOPARD syndrome, a pathogenic mutation being identified in exon 12 of PTPN11 (c.1403C>T; p.Thr468Met). A heterozygous mutation in exon 17 of gene MYBPC3 (p.Gly507Arg) was also detected in these two children; this is a genetic variant of uncertain significance, but predicted to be disease-causing by PolyPhen-2 and Mutation Taster.
Risk for SCD was analyzed in the seven patients with positive phenotype and genotype, three of whom presented no conventional risk factor (Table 3).
Stratification of risk of sudden cardiac death in patients with positive phenotype and genotype.
| Case | Family | Conventional risk factor | Genotype | LVOT obstruction (max. LVOT gradient) | Electrocardiographic risk score |
|---|---|---|---|---|---|
| 1 | I | FH of SCD | - | - | 8 |
| 3 | II | Diastolic IVS >30 mm | - | - | 8 |
| 8 | IV | Syncope; FH of SCD | Compound genotypea | - | 7 |
| 11 | VI | Syncope; diastolic IVS >30 mm; FH of SCD | - | - | 12 |
| 12 | VII | - | - | SAM, exercise-induced intraventricular gradient 100 mmHg | 3 |
| 18 | IX | - | - | - | 4 |
| 19 | X | - | - | - | 3 |
FH: family history; IVS: interventricular septum; LVOT: left ventricular outflow tract; SAM: systolic anterior movement; SCD: sudden cardiac death.
One patient presented a compound genotype, with more than one mutation in a sarcomeric gene.
Four patients had an electrocardiographic risk score of ≥6 (Table 4). Two of the three patients with an electrocardiographic risk score of <6 presented no other risk factors for SCD.
Electrocardiographic risk scores in patients with positive phenotype and genotype.
| ECG parameter | Score | Case 1 | Case 3 | Case 8 | Case 11 | Case 12 | Case 18 | Case 19 |
|---|---|---|---|---|---|---|---|---|
| QRS axis deviation | 1 point | 1 | 0 | 0 | 1 | 0 | 1 | 0 |
| T-wave inversion in limb leadsa | 1 point | 0 | 1 | 1 | 1 | 0 | 0 | 0 |
| T-wave inversion in precordial leadsa | 2 points | 2 | 1 | 1 | 1 | 0 | 0 | 0 |
| ST depression >2 mm | 2 points | 0 | 0 | 1 | 2 | 0 | 0 | 0 |
| Dominant S in V4 | 2 points | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Limb-lead QRS amplitude sum | >7.7 mV=1 point | 0 | 3 | 1 | 3 | 0 | 0 | 0 |
| >10 mV=2 points | ||||||||
| >12 mV=3 points | ||||||||
| 12-lead QRS amplitude-duration sum | >2.2 mV.s=1 point | 3 | 3 | 3 | 3 | 3 | 3 | 3 |
| >2.5 mV.s=2 points | ||||||||
| >3 mV.s=3 points | ||||||||
| QTc >440 ms | 1 point | 1 | 0 | 0 | 1 | 0 | 0 | 0 |
| Total score | 8 | 8 | 7 | 12 | 3 | 4 | 3 |
QTc: corrected QT.
Mean follow-up was 3.5±0.8 years (6 months - 9.5 years).
Patient characteristics at last assessment.
| Case | Family | Age (years) | Phenotype | Symptoms | Complications | Classification |
|---|---|---|---|---|---|---|
| 1 | I | 16 | HCM | 0 | - | Affected |
| 2 | II | 10 | HCM | 0 | - | Affected |
| 3 | II | 19 | HCM | 0 | - | Proband |
| 5 | III | 6 | N | Palpitations | - | At risk |
| 6 | III | 4 | N | 0 | - | At risk |
| 8 | IV | 14 | HCM | 0 | - | Affected |
| 9 | V | 8 | N | 0 | - | At risk |
| 10 | V | 9 | N | 0 | - | At risk |
| 11 | VI | 17 | HCM | 0 | ICD | Affected |
| 12 | VII | 18 | HCM | 0 | - | Proband |
| 13 | VII | 15 | HCM | 0 | - | Affected |
| 16 | IX | 3 | N | 0 | - | At risk |
| 17 | IX | 12 | N | 0 | - | At risk |
| 18 | IX | 7 | HCM | 0 | - | Proband |
| 19 | X | 17 | HCM | 0 | - | Affected |
HCM: hypertrophic cardiomyopathy; ICD: implantable cardioverter-defibrillator; N: normal.
At the end of follow-up, two children with negative phenotype but carrying a mutation in MYBPC3 developed HCM at 10 and 15 years of age (28% penetrance).
All patients diagnosed with HCM at initial assessment still had this diagnosis at the last assessment.
There were no deaths during follow-up. One patient underwent implantation of an ICD as primary prevention following an episode of nonsustained VT on 24-hour Holter monitoring. This patient had three conventional risk factors (syncope, severe LV hypertrophy and a family history of SCD in a paternal aunt diagnosed with HCM) and an electrocardiographic risk score of 12 (Table 3).
DiscussionHCM has an estimated annual incidence of 0.3-0.5 per 100000 children.3,11
Familial HCM in adults is caused by mutations in cardiac sarcomere protein genes in up to 60% of cases,3 but its etiology is more complex and variable in children.
Of 855 children with HCM in the Pediatric Cardiomyopathy Registry, the etiology was known in only 25.8% of cases: 9% were associated with malformation syndromes, 8.7% with inborn errors of metabolism and 7.5% with neuromuscular disorders.12 It is estimated that around a third of children with HCM have familial disease, the result of mutations in genes coding for sarcomeric proteins.9
Genetic testing in familial hypertrophic cardiomyopathyOver 1400 mutations in 11 genes encoding proteins of the myofilaments or Z-disc of sarcomeres have been described.5
A sarcomeric mutation is identified in 50-60% of cases of familial HCM.5 When a pathogenic mutation is identified in a patient, genetic testing is a cost-effective method of family screening, the purpose of which is to detect the presence of or assess the risk of developing the disease in first-degree relatives of the proband, in order to initiate early treatment and stratify the risk of SCD.
The latest ESC and ACCF/AHA guidelines recommend the assessment of child relatives from the age of 10-12 years.3,4 However, in families with early-onset disease, clinical evaluation and genetic testing may be appropriate before this age.3
At present, genetic study is mainly used as an aid in deciding whether to maintain relatives of the proband in clinical and echocardiographic follow-up.3,4
In our sample, five children (25%) presented negative phenotype and genotype and were discharged from regular follow-up. This approach reassures non-affected relatives as to the likelihood of developing the disease and avoids unnecessary consultations.
Children with positive genotype were divided into two groups: those with positive phenotype (n=7, 35%) were classified as affected and those with negative phenotype (n=8, 40%) were classified as at risk.
The ClinVar database and HGMD were used to classify the disease-causing potential of DNA variants, which showed that the mutations identified in eight cases had been previously described as pathogenic. PolyPhen-2 and Mutation Taster were used in cases of previously undescribed mutations (n=3) and of genetic variants of uncertain significance (n=7), to assess their functional effects.
We opted to include patients from family IX in this study despite a genetic diagnosis of LEOPARD syndrome, which in itself would explain the HCM phenotype, since they also presented a heterozygous mutation in exon 17 of gene MYBPC3 (p.Gly507Arg). This is a genetic variant of uncertain significance, likely to be pathogenic according to PolyPhen-2 and Mutation Taster, but the disease-causing potential of this sarcomeric mutation remains to be confirmed.
It was our policy to maintain follow-up of patients with genetic variants of uncertain significance and those with previously undescribed mutations, even in the absence of phenotypic manifestations of the disease, as being at risk.
However, besides serial assessments, the clinical management of carriers of sarcomeric gene mutations with negative phenotype has not been established.3 According to the latest international guidelines, children with positive genotype/negative phenotype should be assessed every 12-18 months, while adults only need to be assessed every 2-5 years.3
Carriers of sarcomeric gene mutations with no phenotypic expression of the disease have a low risk of adverse cardiac events, a recent study reporting an SCD rate of 0.13% per person-year.13
Risk stratification of sudden cardiac deathThe annual risk of SCD in HCM patients is estimated at 1%.13 However, a Danish study carried out between 2000 and 2006 showed a risk of <0.1% per person-year in individuals aged 1-35 years.14
The latest ESC guidelines indicate the following as major risk factors for SCD in children with HCM: very severe LV hypertrophy (defined as maximum LV wall thickness of ≥30 mm or z-score ≥6), unexplained syncope, nonsustained VT, and family history of SCD.3
We opted to stratify risk of SCD in affected relatives only, as recommended in the ACCF/AHA guidelines.4
Besides conventional risk factors, we analyzed other factors described in the literature: LVOT obstruction9 and compound genotype.3,15
We also applied the electrocardiographic risk score proposed by Östman-Smith et al.,9 which was significantly associated with SCD in their population of adults with HCM, with high sensitivity (85%) and specificity (100%). This predictive model assesses the presence of QRS axis deviation, pathological T-wave inversion, ST-segment depression, dominant S in V4, limb-lead QRS amplitude sum, 12-lead QRS amplitude-duration product, and QTc.
It is interesting to note that the only patient in our sample with complications (requiring ICD implantation) had the highest electrocardiographic risk score, as well as three conventional risk factors.
Phenotypic expression of hypertrophic cardiomyopathyThe clinical expression of HCM is determined by a complex interaction between genetic, epigenetic and environmental factors. As with other diseases of autosomal dominant inheritance, HCM shows considerable phenotypic variability, even within the same family.
The following have been suggested as pre-phenotypic manifestations of HCM in carriers of sarcomeric gene mutations: myocardial crypts, elongation of the mitral leaflets, diastolic dysfunction, increased collagen deposition and myocardial fibrosis.3,4
Mutation penetrance of HCM also varies over time and may be substantially delayed, but increases with age, although always to less than 100%.5
A recent study reported an incidence of manifest HCM in carriers of sarcomeric mutations aged under 40 of <0.10% per person-year.13
Studies in children have shown a penetrance of phenotypic expression of 6-31% in carriers of sarcomeric gene mutations after a follow-up of up to 12 years.2,16
In our sample, two phenotype-negative mutation carriers developed HCM after 3.5±0.8 years of follow-up, at age 10 and 15 years. The resulting penetrance rate of 28% is in agreement with the literature.
This highlights the importance of long-term follow-up of carriers of sarcomeric gene mutations, with a view to the eventual development of therapies that may modulate expression of the disease.
Study limitationsThe study has the limitations inherent to its retrospective design and small sample size.
It is important to stress that age-related penetrance probably depends on multiple factors, including gender, race and genotype. Our sample consisted solely of Caucasian children with mutations in four sarcomeric genes (MYBPC3, MYH7, TNNT2 and MYL3).
The small number of complications in our study population does not enable associations to be established with the risk factors identified.
ConclusionWhen a pathogenic mutation is detected in a patient with HCM, genetic testing is an effective means of family screening, as this identifies relatives at risk of developing the disease and those who can be discharged from follow-up.
The penetrance of HCM in child relatives with positive genotype/negative phenotype at initial assessment was 28% after 3.5 years of follow-up. This underlines the need for longitudinal long-term monitoring of sarcomeric gene mutation carriers, irrespective of the presence of a positive phenotype.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Cardoso B, Gomes I, Loureiro P, Trigo C, F. Pinto F. Diagnóstico clínico e genético de miocardiopatia hipertrófica familiar: resultados em cardiologia pediátrica. Rev Port Cardiol. 2017;36:155–165.
