








Turner syndrome is a relatively common genetic disorder of female development, characterized by partial or complete absence of an X chromosome, with a variable clinical presentation. Congenital or acquired cardiovascular disease is highly prevalent and a major cause of early death in this syndrome. The most feared complication is aortic dissection, which can occur at a very young age and requires careful assessment of its risk factors. A systematic literature search identified sixty relevant publications. These were reviewed with regard to the increased risk of cardiovascular disease in women with Turner syndrome, especially in pregnancy. The most common congenital cardiovascular defects are presented and illustrated with appropriate iconography. The current recommendations regarding the screening and monitoring of cardiovascular disease in these patients are discussed.
A síndrome de Turner é uma condição genética relativamente comum, caracterizada pela perda total ou parcial de um cromossoma X, com uma apresentação clínica variável e que afeta o desenvolvimento das mulheres. Nessa síndrome há uma prevalência elevada de doença cardiovascular, congénita e adquirida, que condiciona um risco aumentado de morte prematura. A complicação mais temida é a disseção da aorta, que tende a ocorrer em idades mais precoces, implica uma avaliação cuidada dos seus fatores de risco. A pesquisa sistemática da literatura permitiu encontrar 60 artigos relevantes sobre o tema. Foi feita uma revisão sobre o risco aumentado de doença cardiovascular nas doentes com síndrome de Turner, em particular na gravidez. Foram apresentados e ilustrados com iconografia própria os defeitos cardiovasculares congénitos mais prevalentes. Foram discutidas as recomendações atuais relativas ao rastreio e à vigilância da doença cardiovascular nessas doentes.
Turner syndrome (TS) is a genetic disorder with a characteristic phenotype that occurs as a result of a structurally abnormal or absent X chromosome. TS is the only monosomy compatible with life and affects around 1 in 2000 newborn females.1,2
There are variable Turner karyotypes, such as monosomy X (45,X; the most prevalent karyotype), mosaic karyotype, isochromosome X, ring X chromosome or deletions. In all these cases, significant portions of the X chromosome are deleted.3
The diagnosis of TS requires having key clinical features, including short stature and gonadal failure (the cardinal features) and congenital cardiovascular (CV) defects as well as an abnormal karyotype.2
The most serious consequences of X-chromosome haploinsufficiency involve the CV system. Indeed, CV defects, present in up to 50% of the TS population, are the major cause of premature death and contribute to a standardized mortality ratio three times higher than in the general female population. Therefore, imaging studies (both echocardiography and cardiovascular magnetic resonance [CMR]) are crucial for the timely detection of often subclinical CV disease and should preferably be ordered before the onset of symptomatic and irreversible organ damage.2,4,5
A thorough cardiac examination is recommended for all women with TS every 5 to 10 years, at the time of transition from pediatric to adult care, before attempting pregnancy or if hypertension (HTN) develops.2,6
The aim of this paper is to review both congenital and acquired cardiovascular diseases and the role of clinical imaging in TS CV risk stratification with recommendations for CV screening. Moreover, particular attention will be given to the special risk of CV complications in pregnancy with this syndrome.
MethodsThis review was performed according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines, thus providing a comprehensive framework that objectively assesses quality indicators and risk of bias in the studies included.
All original studies investigating Turner syndrome and cardiovascular risk were eligible for this systematic review. Further selection criteria were: (i) publication date between January 2000 and September 2016, (ii) written in English, (iii) published in a scholarly peer-reviewed journal.
Studies were found by searching the PubMed, EMBASE and Cochrane Library electronic databases, using the following search strategy: “Turner syndrome AND Cardiovascular disease AND Risk factors AND Female”.
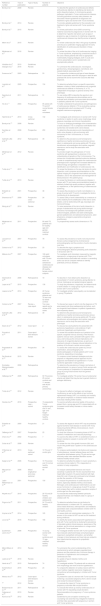
Studies were identified by searching relevant papers via PubMed/MEDLINE (http://www.ncbi.nlm.nih.gov/pubmed), the Cochrane Library and EMBASE using the following search strategy: “Turner syndrome AND Cardiovascular disease AND Risk factors. Finally, reference lists of the studies retrieved were manually searched in order to detect any additional relevant studies. Keywords and combinations of keywords were used to search the electronic databases (Figure 1).
After performing the initial literature searches, each study title and abstract was screened for eligibility by the first author. The full texts of all potentially relevant studies were subsequently retrieved and examined further for eligibility. The PRISMA flow diagram (Figure 1) provides more detailed information regarding the process for selecting studies. Sixty articles were included in this review and the information from the studies included was then analyzed and recorded. Table 1 shows a summary of the literature consulted.
Summary of the literature consulted.
| Reference number | Year of publication | Type of study | Number of patients | Objective |
|---|---|---|---|---|
| Bondy et al.1 | 2008 | Review | To describe the spectrum of cardiovascular defects with particular attention to identifying risk factors for aortic dissection/rupture. X-chromosome genetic pathways implicated in Turner cardiovascular disease, including premature coronary artery disease, are discussed. Recent guidelines for diagnosis and treatment of girls and women with TS are reviewed. | |
| Bondy et al.2 | 2014 | Review | To provide updated guidelines for the evaluation and treatment of girls and women with Turner syndrome (TS). | |
| Bondy et al.3 | 2015 | Review | To review publications since 2000 concerning maternal outcomes for pregnancies in women with TS to determine if specific risk factors such as type of pregnancy, age or presence of underlying congenital cardiovascular disease may identify women at special risk. | |
| Marin et al.4 | 2015 | Review | To review cardiovascular risks in this group of patients and discuss a systematic imaging approach for early identification of cardiovascular disorders in these patients. | |
| Mortensen et al.5 | 2016 | Review | To review the state of the art of cardiovascular imaging in Turner syndrome, emphasizing unresolved issues in the care of these patients with links to appropriate multimodality imaging strategies, both in acute and chronic presentations and in symptomatic and asymptomatic patients. | |
| Hiratzka et al.6 | 2010 | Guidelines | ||
| Granger et al.7 | 2016 | Review | To summarize the cardiovascular, neurological, genitourinary, otolaryngological, craniofacial, and skeletal defects associated with TS. | |
| Surerus et al.8 | 2003 | Retrospective | 53 | To compare the incidence and type of heart disease found in association with 45X karyotype in fetal life with postnatal life and to examine the outcome after fetal diagnosis. |
| Loscalzo et al.9 | 2005 | Prospective | 134 | To determine whether there is a specific association between a history of fetal lymphedema and congenital cardiovascular defects in monosomy X, or TS, independent of karyotype or general severity of the phenotype. |
| Spectrum et al.10 | 2011 | Retrospective | 51 | To use cardiac MRI to describe the spectrum and frequency of cardiovascular abnormalities and to evaluate aortic dilatation and associated abnormalities in pediatric patients with Turner syndrome. |
| Ho et al.11 | 2004 | Prospective | 85 adults with TS and 27 normal female adult volunteers | To better understand the prevalence and pathogenesis of cardiovascular defects in TS by evaluating a group of asymptomatic adult volunteers with TS using magnetic resonance angiography. |
| Hjerrild et al.12 | 2010 | Cross-sectional | 102 | To investigate aortic dimensions in women with Turner syndrome in relation to aortic valve morphology, blood pressure, karyotype and clinical characteristics. |
| Bondy et al.13 | 2008 | Review | To focus on emerging knowledge of the characteristics of aortic disease in TS in comparison with Marfan-like syndromes and isolated aortic valve disease. | |
| Sachdev et al.14 | 2008 | Prospective | 253 | To determine the prevalence and characteristics of aortic valve disease in girls and women with monosomy for the X chromosome, or Turner syndrome. |
| Gutmark-Little et al.15 | 2012 | Retrospective | 39 | To determine the prevalence and hemodynamic significance of partial anomalous pulmonary venous return in adolescents and young adults with TS using transthoracic echocardiogram and cardiac magnetic resonance, and to study the association with phenotype. |
| Mortensen et al.16 | 2012 | Review | To provide insight into pathogenesis of Turner syndrome with perspectives to advances in the understanding of genetics of the X-chromosome. The review also incorporates important endocrine features, in order to comprehensively explain the cardiovascular phenotype and to highlight how increased attention to endocrinology and genetics is important in the identification and modification of cardiovascular risk. | |
| Turtle et al.17 | 2015 | Review | To review the literature on the key risk factors for aortic dissection, and to propose recommendations on the optimal imaging for congenital heart defects and aortic dilatation, and on the assessment and management of blood pressure in this patient group. | |
| Turtle et al.18 | 2013 | Review | To review the literature on cardiovascular disease in women with TS and make recommendations based on relatively limited high-quality evidence, together with our experience, on the optimal timing of cardiovascular screening. | |
| Elsheikh et al.19 | 2001 | Prospective | 38 | To assess the prevalence of aortic root dilatation in a group of women with TS and to investigate the factors contributing to its development. |
| Sharma et al.20 | 2009 | Prospective cross-sectional | 24 | To assess utility of aortic distensibility as a measure of aortic elasticity for the stratification of the risk for aortic dilation, and its relationship with age, karyotype and hormonal therapy. |
| Wong et al.21 | 2014 | Review | To raise awareness of the risk of aortic dissection in groups of adolescents and young adults amongst clinicians of all backgrounds (pediatric and adult endocrinologists, pediatric and adult cardiologists, general pediatricians and adolescent physicians, adult physicians, emergency specialists and general practitioners). | |
| Mortensen et al.22 | 2011 | Prospective | 80 adult TS patients and 67 healthy-age and gender-matched controls | To prospectively assess aortic dimensions in TS. |
| Lanzarini et al.23 | 2007 | Prospective | 78 | To assess the prevalence of aortic root and proximal thoracic aorta enlargement by 2-dimensional echocardiography and the evolution of aortic diameters over time. |
| Carlson et al.24 | 2012 | Prospective | 20 | To describe the signs, symptoms and echocardiographic features preceding aortic dissection in 20 women with TS. |
| Matura et al.25 | 2007 | Prospective | 166 adult volunteers with TS who were not selected for cardiovascular disease and 26 healthy female control subjects. | To investigate aortic diameters measured by magnetic resonance in a large group of women with TS to elucidate factors associated with variation in ascending aortic size and to compare methods to identify potentially ominous dilatation. |
| Gravhoolt et al.26 | 2006 | Retrospective | 18 | To describe in more detail aortic dissection as encountered in Turner's syndrome, giving attention to clinical, histological and epidemiological aspects. |
| Lopez et al.27 | 2015 | Prospective | 138 | To study the prevalence, magnitude, and determinants of aortic dilation in a large group of girls and young women with Turner syndrome. |
| Lawson et al.28 | 2014 | Prospective | 49 Turner syndrome patients and 76 lean and 52 obese controls | To investigate whether vasculopathy can be detected in young TS patients. |
| Carlson et al.29 | 2007 | Review + case report | Two case reports and a review of 85 cases | To discuss two cases in which only the diagnosis of TS was present and other predisposing risk factors, such as congenital heart disease or systemic hypertension, were expressly absent. |
| Gutmark-Little et al.30 | 2012 | Retrospective | 39 | To determine the prevalence and hemodynamic significance of partial anomalous pulmonary venous return in adolescents and young adults with TS using transthoracic echocardiogram and cardiac magnetic resonance, and to study the association with phenotype. |
| Sears et al.31 | 2012 | Case report | 2 | To discuss two adult patients who presented with pulmonary hypertension, and evidence of right ventricular hypertrophy and dysfunction. |
| Povoski et al.32 | 2011 | Case report with literature review | 1 | To review the available literature as it pertains to the specific congenital venous anomaly of the thoracic systemic venous return, persistent left superior vena cava, and to discuss the clinical implications and relevance of congenital aberrancies, as well as of treatment-induced or disease-induced alterations in the anatomy of the thoracic central venous system. |
| Proprawski et al.33 | 2009 | Prospective | 34 | To assess the cardiovascular system by clinical examination, echocardiography and electrocardiography in female patients with Turner syndrome, depending on karyotype. |
| De Groote et al.34 | 2015 | Review | To compare current treatment strategies and also propose an integrated practical approach for the diagnosis and treatment of hypertension in Turner syndrome applicable in daily practice. | |
| Kozlowka-Wojciechowska et al.35 | 2006 | Retrospective | To determine the prevalence of classic risk factors of atherosclerosis in a group of young Polish women with Turner syndrome in comparison to a representative group of young Polish women from the general population. | |
| Ostberg et al.36 | 2005 | Retrospective | 93 TS women, 25 normal controls and 11 women with 46,XX primary amenorrhea | To investigate whether TS women have a fundamental arterial wall defect that may be due to genetic factors or estrogen deficiency. |
| Trolle et al.37 | 2012 | Review | To discuss the effects of estrogen and androgen insufficiency as well as the effects of sex hormone replacement therapy (HRT) on morbidity and mortality with special emphasis on evidence-based research and areas needing further studies. | |
| Groote et al.38 | 2016 | Prospective case – control | 15 prepubertal Turner syndrome girls and 31 sex-, age- and height-matched healthy controls | To examine whether abnormal rigidity of the aorta is present from a young age and if it is associated with an increased carotid artery diameter or intima-media thickness. |
| Elsheikh et al.39 | 2000 | Prospective | 21 | To assess the degree to which HRT may be protective against ischemic heart disease in women with TS by measuring arterial stiffness, blood pressure, insulin sensitivity and lipids. |
| Ostberg et al.40 | 2007 | Prospective | 25 | To study the influence of increasing doses of HRT on markers of metabolism and vascular physiology. |
| Chan et al.41 | 2002 | Prospective | 7 | To examine the effect of HRT on endothelial function of forearm resistance vessels in women with Turner's syndrome. |
| Colao et al.42 | 2008 | Review | To review the role of the growth hormone (GH) and insulin-like growth factor I system on the cardiovascular system. | |
| O’Gorman et al.43 | 2013 | Cross-sectional cohort | 19 TS and 17 control girls | To compare cardiometabolic risk factors and measures of subcutaneous, visceral adipose tissue and intra-myocellular lipid between young TS girls and an age- and body mass index-standard deviation scores-matched healthy female cohort. |
| Salgin et al.44 | 2006 | Prospective | 16 TS women and 16 normal healthy women | To determine whether the defect in insulin sensitivity is a primary intrinsic defect in TS or dependent on variation in body composition. |
| Mazzanti et al.45 | 2006 | Mixed longitudinal and cross-sectional | 46 | To analyze carbohydrate tolerance and insulin sensitivity in patients with TS who reached their final height after long-term (10 years) treatment with high-dose GH, and in those who continued to be followed-up after the cessation of GH therapy. |
| Ladin-Wilhelmsen et al.46 | 2001 | Prospective | 100 | To analyze the prevalence of cardiovascular risk factors, especially diabetes mellitus, blood lipids, fibrinogen, and cardiovascular malformations, as well as sex hormones in a large group of adult TS women and compare them with a random population sample of similar age from the same region. |
| Akyrek et al.47 | 2015 | Prospective | 29 TS and 29 healthy children and adolescents | To evaluate the relationship between periaortic adipose thickness and metabolic data in a group of children with TS. |
| Pirgon et al.48 | 2011 | Prospective | 24 TS and 24 healthy children | To determine whether there are risk factors for atherosclerosis in children with TS, carotid artery intima-media thickness, anthropometric and metabolic parameters were compared between children with TS and healthy controls. |
| Irzyniec et al.49 | 2014 | Prospective | 165 | To assess lipid and carbohydrate metabolism in TS women in the context of HRT as well as GH treatment during childhood. |
| Los et al.50 | 2016 | Prospective | 168 | To identify the factors associated with hypertension in girls with Turner syndrome by studying the prevalence of elevated systemic blood pressure, awareness of the problem, and its clinical associations in a large cohort of girls with Turner syndrome |
| Zuckerman-Levin et al.51 | 2006 | Prospective | 10 young women with Turner syndrome and 10 age-matched healthy women | To investigate whether women with Turner syndrome have reduced catecholaminergic and physiological response to sympathetic stimulation, and whether changes in blood pressure and heart rate are related to their catecholamine response to sympathetic stimulation. |
| Maric-Bilkan et al.52 | 2014 | Review | To summarize the current understanding of the mechanisms by which estrogens regulate blood pressure and the potential use of hormone therapy in prevention of hypertension and consequent cardiovascular risk. | |
| Lastra et al.53 | 2016 | Review | An update of type 2 diabetes mellitus and hypertension. | |
| Herck et al.54 | 2015 | Retrospective | 74 | To investigate whether TS patients with an abnormal arch morphology are more at risk for hypertension. |
| Somerville et al.55 | 2016 | Prospective | 24 | To compare the detection of cardiac lesions with the use of cardiac magnetic resonance imaging and conventional echocardiography in children with Turner syndrome. |
| Nwosu et al.56 | 2012 | Case report with literature review | 1 | To present the case of a woman with Turner syndrome achieving a successful pregnancy from a donor oocyte and review the relevant literature. |
| 57 | 2012 | Review | To evaluate the impact of Turner syndrome on pregnancy outcomes after oocyte donation. | |
| Boissonnas et al.,58 | 2011 | Retrospective | 25 | To evaluate the follow-up (mainly cardiovascular) of women with TS requesting oocyte donation. |
| Cabanes et al.59 | 2010 | Review | Recommendations for pregnancy in Turner syndrome patients. | |
| Karnis et al.60 | 2012 | Review | Guidelines for pre-conception screening and counseling now exist that may mitigate the maternal and fetal risks associated with pregnancy in women with Turner syndrome. |
Variations in the CV anatomy of patients with TS, the frequencies of which are summarized in Table 2, are major factors in their reduced life expectancy.7
Frequency of the main congenital cardiovascular anomalies in TS.
| Congenital heart defect | Frequency |
|---|---|
| Aortic anomalies: | |
| Elongated transverse arch | 31.4%,10 49%11 |
| Aortic dilatation | 23%12 |
| Aortic dissection | 100-fold increased risk13 |
| Coarctation of the aorta | 12%,11 15.7%10 |
| Valvular anomaly: | |
| Bicuspid aortic valve | 25%,12 30%,14 39.2%10 |
| Venous anomalies: | |
| Partial anomalous venous connection | 13%,11 15.7%,1018%15 |
| Persistent left superior vena cava | 13%11 |
| Arch vessel anomaly: | |
| Aberrant right subclavian artery | 8%11 |
Major defects in cardiac and aortic development during fetal life are associated with miscarriage in most cases of fetuses with 45X karyotype. Fetuses with CV failure almost always demonstrate obstructed jugular lymphatics with increased nuchal translucency or nuchal cystic hygromas, the typical intrauterine presentation of TS. The residual postnatal webbing of the neck predicts CV defects. This association suggests a pathogenetic connection between fetal lymphatic obstruction and defective CV development due to the compression or obstruction of the outflow tracts.2,8,9
However, this is just a theory regarding a pathogenetic mechanism for congenital CV defects, because the real causes are currently unknown.1
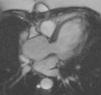
A. Valvular anomalyA.1. Bicuspid aortic valveA bicuspid aortic valve (BAV) is a congenital valvular anomaly found in approximately 30% of TS patients and it is not often clinically apparent.14
Identifying a BAV in asymptomatic individuals is important because they are at increased risk of infective endocarditis, hemodynamically significant stenosis (promoted by accelerated valve calcification), valve regurgitation (secondary to poor leaflet coaptation) and aortic aneurysm (due to the vicious cycle of increased stroke volume that promotes aortic root dilatation). For that reason, the anomaly requires regular medical evaluation and possibly surgery to prevent aortic dissection or rupture.14,16
Indeed, the aortic valve leaflet must be clearly visualized and echocardiography is the primary test for BAV that has been reported to be quite sensitive in diagnosing this anomaly in 89% of women with TS. However, echocardiography can be inadequate to view the aortic valve and, in these cases, CMR should be the next approach.4,14
In almost all the cases, BAV results from fusion of the right and left coronary leaflets, while the fused right coronary and non-coronary leaflets variant is much less common4,14 (Figure 2).
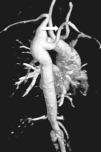
B. Aortic abnormalitiesB.1. Coarctation of the aorta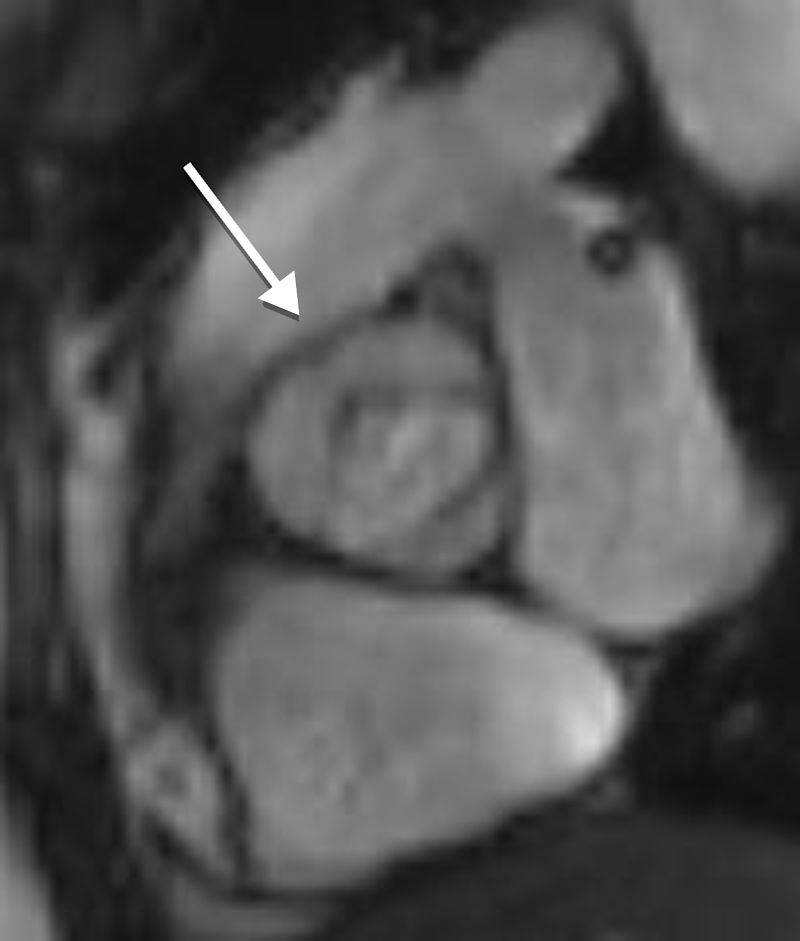
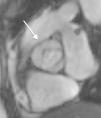
Coarctation of the aorta (CoA) affects around 12% of women with TS11 and is usually diagnosed in infancy, often with congestive heart failure in critical cases17 (Figure 3).
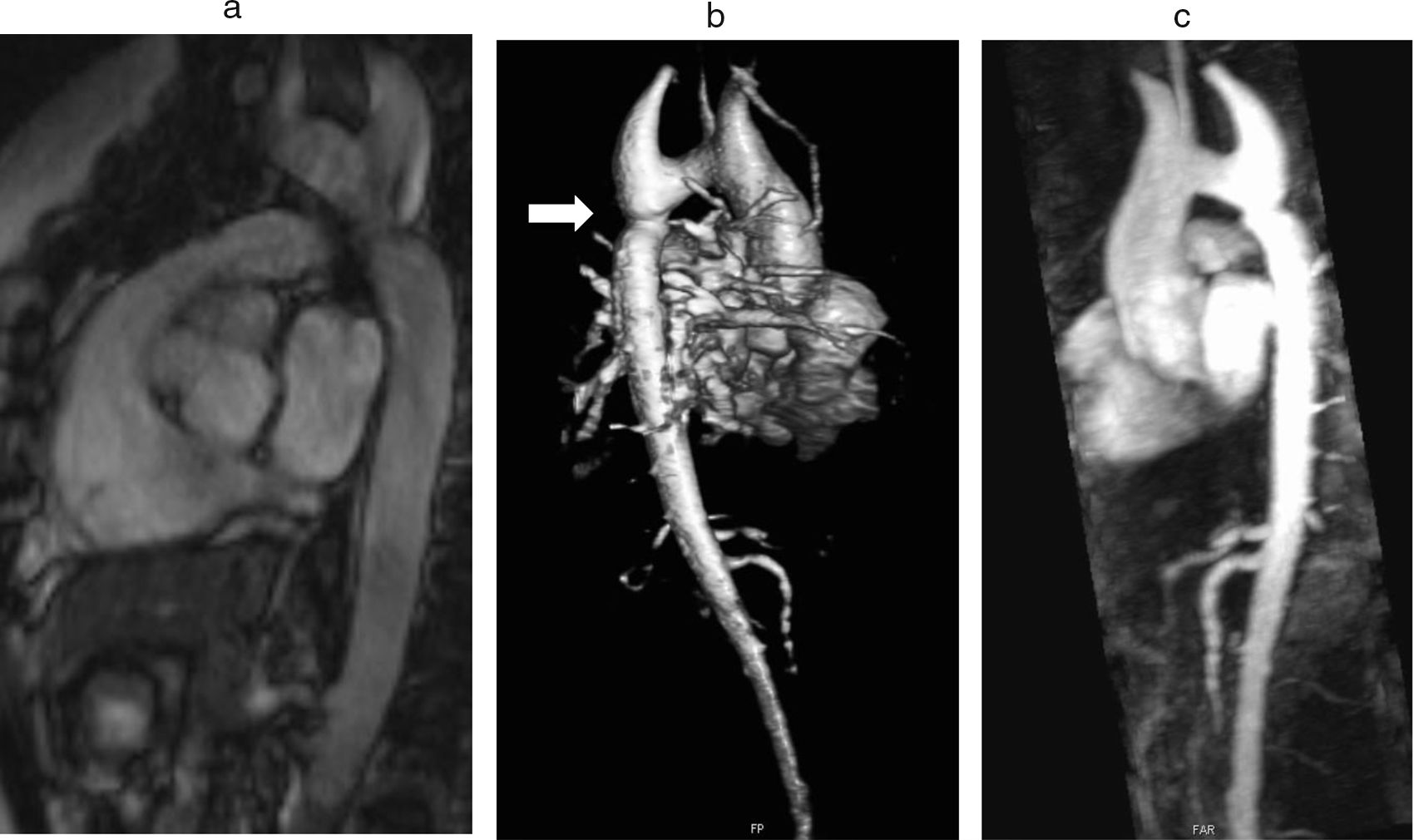
Coarctation of the aorta (white arrow) in a 24-year-old TS woman in different projections: (a) sagittal-oblique cine magnetic resonance image, (b) volume-rendered magnetic resonance aortography (posterior-oblique view), (c) magnetic resonance aortography maximum intensity projection (sagittal-oblique view).
Indeed, in contrast to BAV, which is detected by screening, CoA is usually diagnosed based on clinical grounds – HTN and brachial-femoral delay are common features. However, since many cases are detected later in life, any woman with suspected CoA should have a CMR or computed tomography (CT) angiography with three-dimensional reconstruction of the thoracic aorta.1,11
Concentric narrowing of the aortic lumen leads to the development of pressure gradients across the lesion, with the possibility of arterial collaterals becoming a detour for oxygenated blood traveling to the lower segments of the body.7
If left untreated, complications such as HTN, congestive heart failure, aortic dissection and aortic rupture may occur. Surgical intervention is required when peak-to-peak coarctation gradient is ≥20 mmHg, or <20 mmHg with radiological evidence of significant collateral flow. Even after surgical repair, there is an increased risk of HTN, coronary artery disease (CAD), cerebrovascular disease, aortic dissection and restenosis.18
B.2. Elongated transverse archElongated transverse arch (ETA) is a silent anomaly, detected by CMR, defined as an increased distance between the origin of the left common carotid and left subclavian arteries, with flattening of the arch and kinking along the inferior curvature. This distinct anatomy, sometimes referred to as “pseudocoarctation”, is embryologically similar to coarctation and may predict aortic complications, such as aortic dilation, aortic dissection or progression to overt coarctation.1,11 However, ETA differs from true coarctation in that there is no true luminal narrowing, nor are there pressure gradients or collateral circulation.7
ETA has been significantly associated with BAV, CoA and also with aortic sinus dilatation.10
B.3. Aortic dilatationAortic dilatation is reported in 23% of women with TS.12
Risk factors for this and, secondarily, dissection include HTN, BAV, CoA, 45,X karyotype and pregnancy. Aortic dilatation may less commonly occur alone in approximately 5% of women with TS. This indicates that all women with TS should be periodically monitored.5,19–21
In TS patients with enlargement of the ascending aorta, a general aortopathy is present, which is accelerated by the presence of a BAV.22
It has been suggested that patients with an enlarged aortic root should undergo echocardiographic evaluation every year and that patients with normal aortic root dimensions should be evaluated every 2 to 3 years.23
Aortic diameter, corrected for age and body size, is accurate for predicting aortic events, and is the principal risk marker for aortic dissection.1,5 An ascending to descending aortic diameter ratio above 1.5 signifies ascending aorta dilatation if descending aorta diameter is normal. There is another alternative to identifying aortic dilatation that correlates more closely with aortic diameter, because it adjusts aortic dimensions for body surface area (BSA); this is important due to the relatively short stature of patients with TS. This latter method involves estimating the aortic size index (ASI): aortic diameter (at the ascending aorta or at the site with the largest dimension)/BSA. It has been proposed that an ASI ≥2 cm/m2 identifies those who require close monitoring and an ASI ≥2.5 cm/m2 (as in Figure 4) requires aortic surgery to prevent aortic dissection.4,21,24,25
B.4. Aortic dissection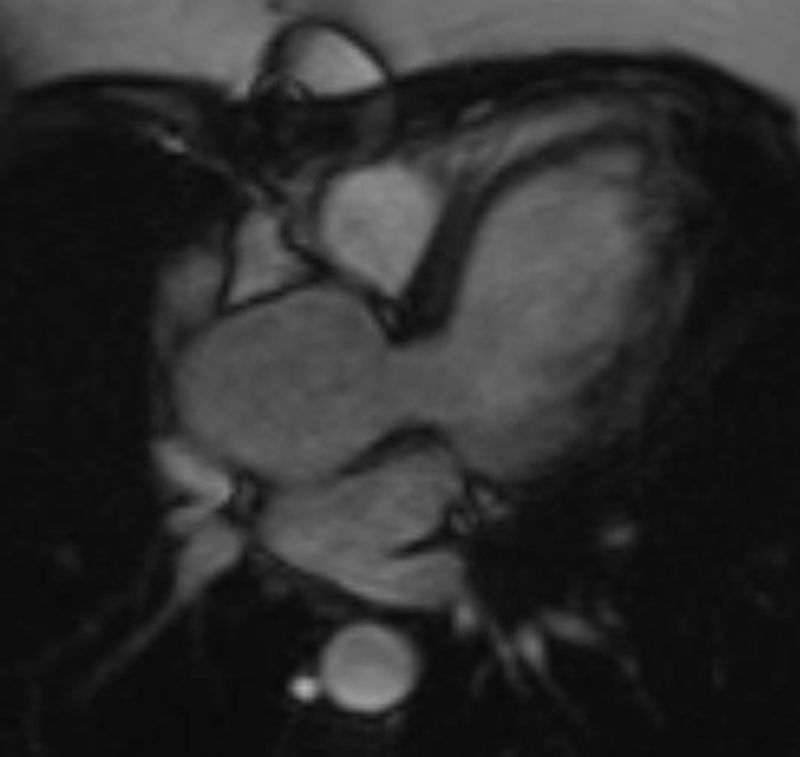
Although rare, aortic dissection is a major concern in TS. It has an estimated incidence of 40 cases per 10000 patients, but is often fatal. It occurs at a much earlier age; in the general population, the peak incidence of dissection occurs between ages 50 and 80 and in TS population median age is 35, with higher incidence rates at ages 20–29 and 30–39.2,26
Up to 90% of aortic dissections have predisposing CV risk factors (BAV, aortic dilatation and CoA) that can be identified with CMR imaging. HTN and pregnancy are associated with a higher risk of dissection, the former due to an association with increased aortic root diameter and the latter due to physiological adaptation, which imposes a higher cardiac workload.4,19
However, up to 25% of cases have no apparent risk factors other than TS itself, suggesting that TS phenotype may also include a primary aortopathy, which can be present as early as 9 years of age.18,26–28
Indeed, histological evidence of cystic medial necrosis in aortic tissue taken from patients with BAV, Marfan syndrome and TS indicates a shared etiology despite genetically diverse backgrounds.29
Aortic dissection is sometimes overlooked at onset because it could include apparently minor complaints such as abdominal pain, heartburn, back or shoulder pain or a voice change due to traction of the recurrent laryngeal nerve. Therefore, patients should be advised to go to the emergency department soon after experiencing sustained chest pain (>30min), regardless of the severity of symptoms.17,29
C. Venous anomaliesC.1. Partial anomalous pulmonary venous connectionIn TS, partial anomalous pulmonary venous connection may be right-sided, which is the most typical in the general population. Nevertheless, it frequently involves the left upper pulmonary vein, which makes echocardiographic detection more challenging.2,30
When the pulmonary veins connect to the right atrium or with one of its venous tributaries, blood from the pulmonary circulation returns to the right atrium and this may become clinically significant in adult life. Indeed, if sufficiently severe, pulmonary HTN and right ventricular volume overload can lead to congestive heart failure, which has been related to death in middle-aged TS patients secondary to undiagnosed partial anomalous pulmonary venous return lesions. Echocardiography and ideally CMR imaging should therefore be performed to assess pulmonary vein drainage patterns.30,31
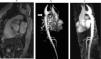
C.2. Persistent left superior vena cavaThe venous return from a persistent left superior vena cava (PLSVC; Figure 5) has a prevalence of around 13%. In approximately 80% to 92% of cases, the PLSVC drains into the right atrium via the coronary sinus, resulting in no hemodynamic consequences. However, in about 10% to 20% of cases, it drains into the left atrium and results in venous blood returning to the left atrium.11,32
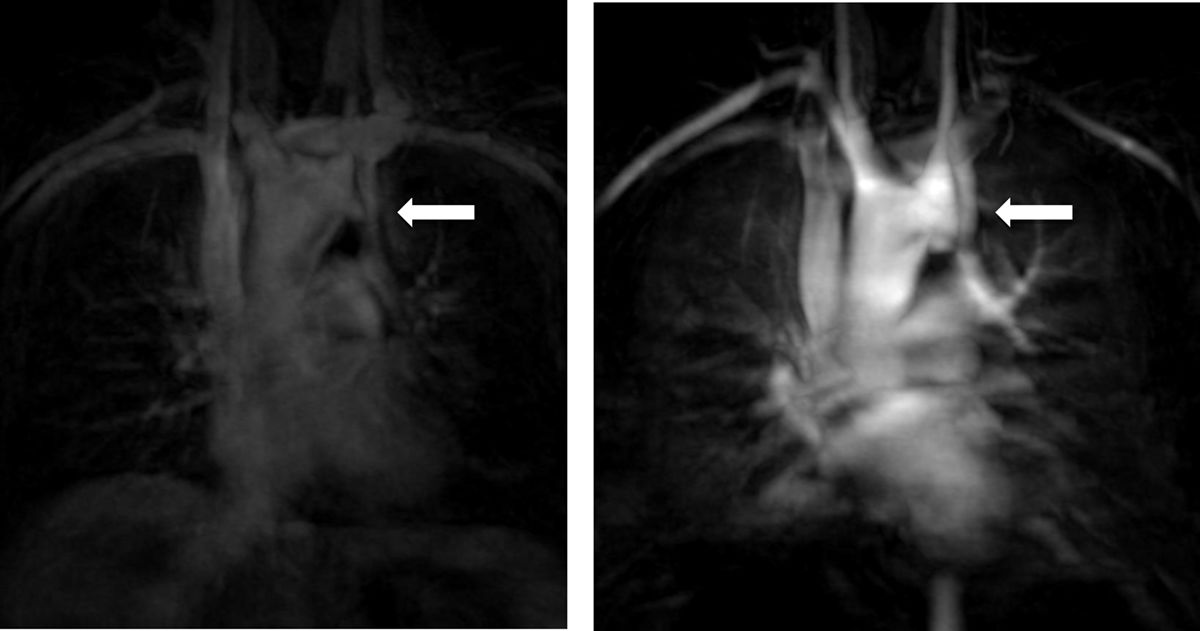
It is important to always report this irregular anatomy, as it needs to be taken into account during interventional procedures.32
D. Other congenital cardiovascular disordersCardiac conduction defects are also common, caused by left ventricular hypertrophy, myocardial ischemia, previous myocardial infarction and congenital cardiac malformations.33
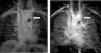
Other associated lesions can be found in TS, such as ventricular and atrial septal defects, hypoplastic left heart syndrome, single ventricle, mitral valve abnormalities, coronary artery abnormalities and aberrant right subclavian artery4 (Figure 6).
Cardiovascular risk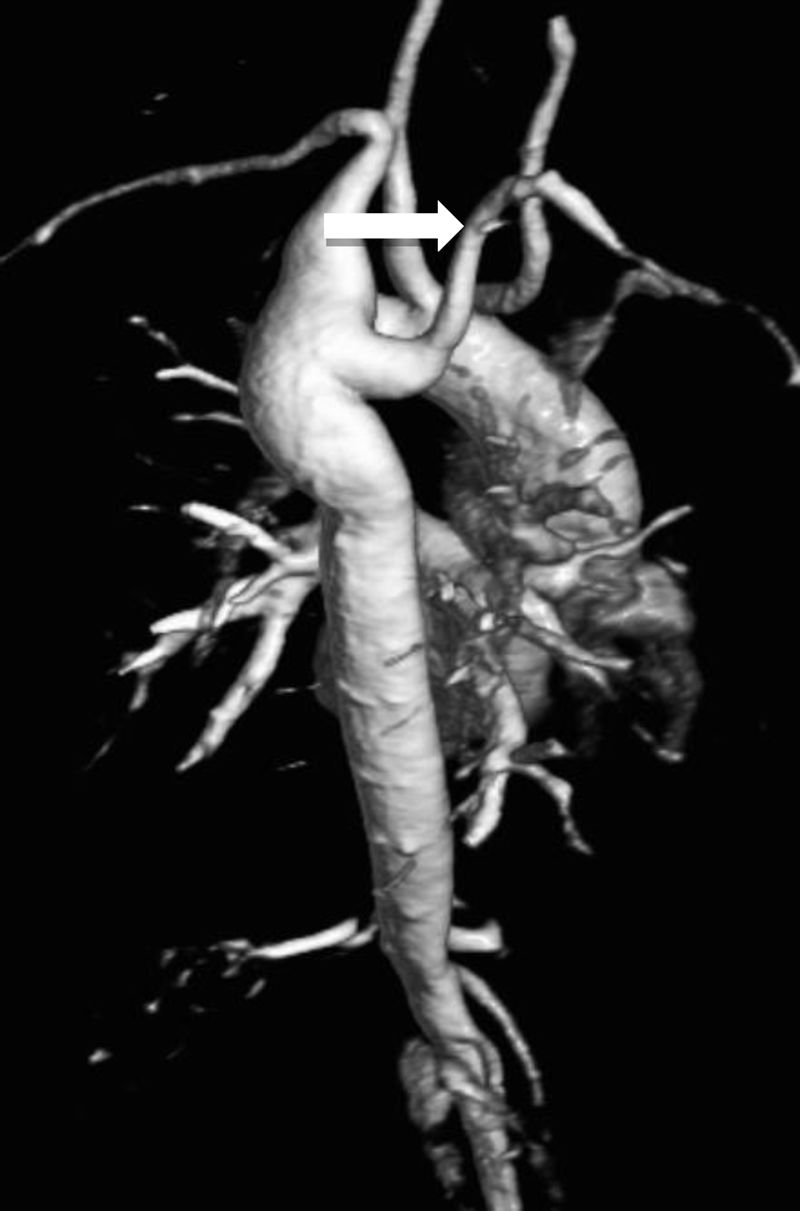
In addition to potentially harmful CV malformations, women with TS display evidence of adverse CV risk profiles that expose them to greater risk of CAD with a relative risk (RR) of 2-11, cerebrovascular disease with a RR of 2-7, HTN and types 1 and 2 diabetes mellitus.16,18,34,35
The precise mechanisms of increased CV risk in TS are unclear, but estrogen deficiencies (ensuing from premature ovarian failure due to gonadal dysgenesis) and haploinsufficiency of the X chromosome seem to be major factors that distinguish this metabolic phenotype.35–37
A. Specific aspects of vascular structure and physiologyWomen with TS have greater intima-media thickness and conduit artery diameters than normal controls.36 The stiffness of the ascending aorta is also increased in these patients.38
Estrogen deficiency may contribute to this, as hormone replacement therapy (HRT) appears to reduce aortic stiffness,39 decrease intima-media thickness40 and improve endothelial function.41 Indeed, gonadal dysgenesis may be the key determinant of neointimal hyperplasia in these subjects, because it has been shown that increasing doses of HRT results in a reduction in carotid intima-media thickness in young hypogonadal women, which raises the possibility that exogenous estrogen may be cardioprotective36,40 (Table 3).
| Estrogen action | Consequence |
|---|---|
| Stimulation of the release of nitric oxide by vascular endothelial cellules | Inhibition of platelet aggregation and monocyte adhesion to the vascular wall |
| Inhibition of superoxide anion production, increasing nitric oxide bioavailability | Vasodilatation (also induced by bradykinin through the bradykinin B2 receptors) Inhibition of vascular smooth muscle proliferation |
Relative resistance to growth hormone (GH) effects and an imbalance in the GH/insulin-like growth factor I axis could also occur, which may have a negative impact on cardiovascular prognosis. In fact, GH replacement (the accepted treatment for the short stature of TS girls) decreases intima-media thickness at major arteries and improves endothelial dysfunction, which are considered surrogate markers of atherosclerosis.16,42
B. Glucose metabolism, body composition and physical activityTS girls have a greater risk of developing diabetes, which is related to both the impaired fasting glucose and the impaired glucose tolerance detected among these girls.43 Moreover, those who have normal fasting glucose concentrations are more insulin resistant than controls.44
The effect of HRT on insulin sensitivity remains unclear, but insulin resistance is a karyotype-dependent factor.44 Furthermore, GH therapy has been shown to reduce insulin sensitivity, antagonizing the effects of insulin on glucose and lipid metabolism via GH receptors. Thus, high GH levels induce peripheral and hepatic insulin resistance. However, patients with TS develop an adaptation to long periods of GH therapy, so insulin sensitivity increases. Upon therapy cessation, insulin sensitivity also increases and returns to pre-treatment levels.45
Additionally, compared to normal controls, TS patients have a greater waist circumference and higher absolute body mass index, which are associated with an increased risk of type 2 diabetes and metabolic dysfunction.43 They also tend to be less physically active, thereby contributing to the increased frequency of obesity.18,46
C. Lipid profileTS patients have an abnormal lipid profile with higher total cholesterol levels, which contributes to the onset of atherosclerosis and also confers an increased risk for CV disease.43,44,47,48
A dose-related relationship between HRT and high-density lipoprotein concentrations has been observed,40 elucidating the benefits of HRT on CV outcomes by improving the lipid profile. Moreover, the impact of estrogens on lipid metabolism is also determined by diminishing lipid oxidation and increasing triglyceride synthesis. However, in one study, the application of HRT in women with TS had no effect on lipid metabolism.49
D. HypertensionHTN can be present in TS patients of any age and has been reported in around 42% of patients. As TS alone carries with it an increased intrinsic risk for HTN, related to the morbidity and mortality of the syndrome, HTN should be carefully diagnosed and treated.34,50
HTN in TS is commonly seen in the absence of cardiac or renal malformations and its pathophysiology is poorly understood and complex.19 Some of the mechanisms that seem to be involved in HTN are increased set risk factors for HTN which are often present in TS, such as obesity, type-1 and type-2 diabetes, dyslipidemia, oxidative stress and inflammation, impaired insulin-mediated vasodilatation and abnormal sodium processing by the kidneys. The role of the inappropriate activation of the renin-angiotensin-aldosterone (RAA) system remains uncertain, but overactivation of the sympathetic nervous system likely plays a major part.34 Estrogen deficiency contributes to blood-pressure (BP) imbalance, because estrogens regulate vascular function by modulating some biological cascades, such as the RAA and endothelin systems. They also have an antioxidant effect and a positive correlation with renal function.34,51–53
Aortic root dilatation is closely dependent on BP, suggesting that rigorous treatment of HTN may reduce the risk of aortic dilatation progression. In addition to that, a significant association between abnormal aortic arch morphology and HTN has been demonstrated.19,54
CoA, even if repaired in the neonatal period, is also a significant risk factor for HTN in TS, suggesting that prenatal or early postnatal hypoperfusion of the kidneys may play a pathogenic role. Haploinsufficiency of critical genes on the X chromosome may be related to the established TS vasculopathy.50
There is no evidence to support specific HTN treatments in TS, and the choice of therapeutic agent should follow general guidelines when BP reduction is the primary objective.16 Target BP should be less than 135/85 mmHg, and in those who have congenital heart defects, such as BAV, CoA and dilatation of the aorta (ASI ≥2.0 cm/m2), should be less than 130/80 mmHg.34
Recommendations for cardiovascular risk screeningAwareness of considerable CV risk requires effective preventive medicine from the earliest years of life.35 Therefore, an appropriate and rigorous CV evaluation with the spectrum of CV issues encountered in TS should be performed (Table 4). The best cardiac imaging modalities for visualizing each of the congenital CV anomalies are shown in Table 5.
Screening and monitoring algorithm for cardiovascular imaging in TS girls and women. Adapted from Marin et al.4
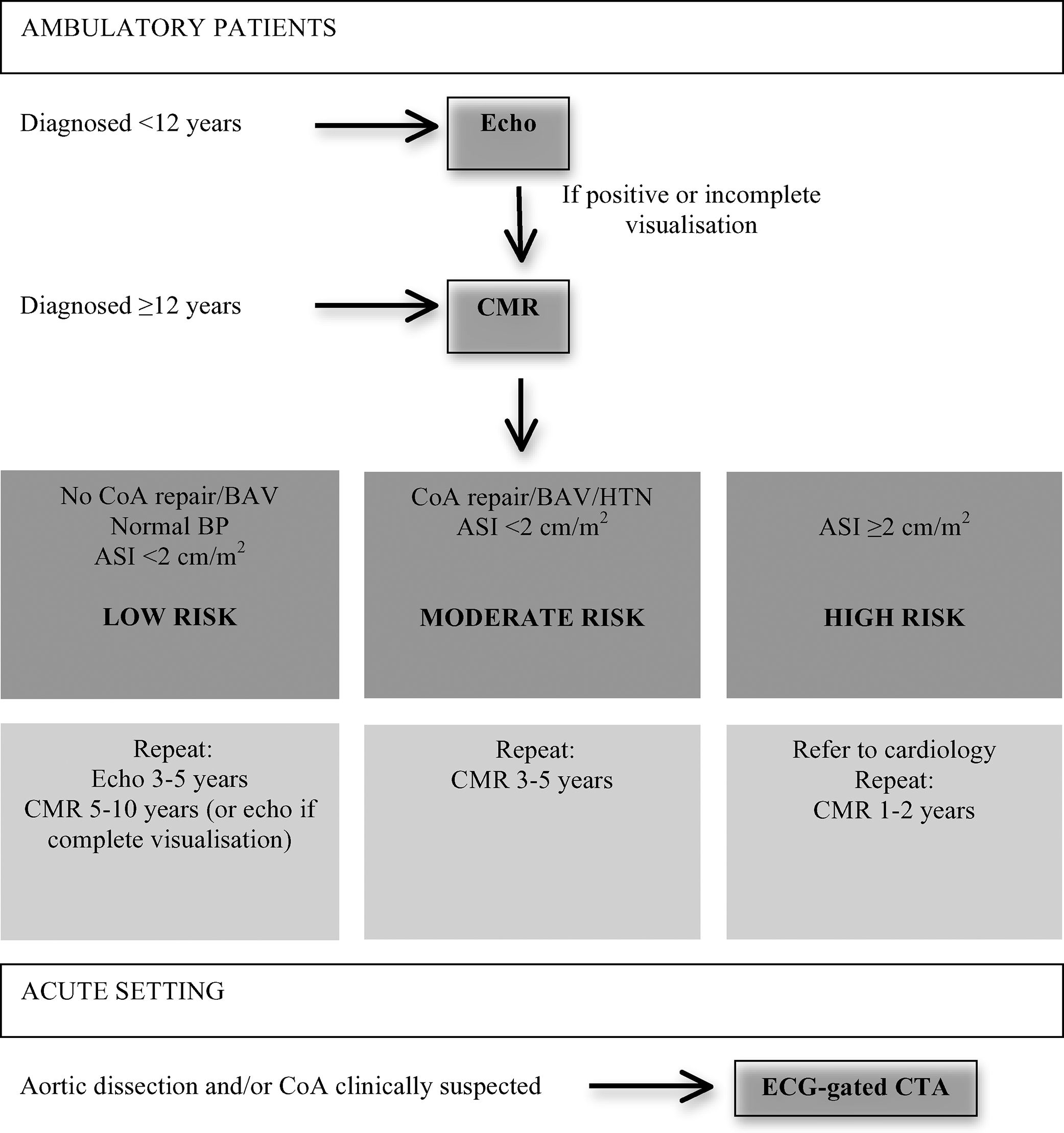
ASI: aortic size index; BAV: bicuspid aortic valve; CMR: cardiovascular magnetic resonance; CoA: coarctation of the aorta; Echo: echocardiography; ECG-gated CTA: electrocardiogram-gated computed-tomography aortography; HTN: hypertension
Type of imaging modality used for cardiovascular screening. Adapted from Wong et al.21
| Congenital cardiovascular defect | Imaging modality |
|---|---|
| Vascular anomalies | |
| Coarctation of the aorta | CMR may be superior, especially in adults |
| Elongated transverse arch | |
| Dilatation of the aorta | Echocardiography may be appropriate for assessment of aortic dimensions if adequate views are obtained CMR if doubts regarding echocardiography measurements exist |
| Valvular anomaly | |
| Bicuspid aortic valve | TTE modality of choice for assessment of valve anatomy and function TEE/CMR when ultrasound windows are limited in older patients |
CMR: cardiovascular magnetic resonance; TTE: transthoracic echocardiography; TEE: transesophageal echocardiography
At diagnosis, all infants and children with TS should undergo comprehensive CV evaluation, including clinical examination, electrocardiogram (ECG) to detect conduction abnormalities, BP measurement and echocardiography, which are currently the standard of care to assess the cardiac anatomy of girls with TS.2,55
CMR is recommended in TS children from the age of 12 (when it can be performed without sedation), even if no cardiac anomalies are detected on echocardiography. Indeed, CMR is the gold-standard imaging test for the diagnosis and monitoring of morphological anomalies of the thoracic aorta in TS, since echocardiography underestimates the size of both the ascending and the descending aortas.5,18
For outpatients, alternating echocardiography and CMR may be helpful. Aortic diameter in TS patients with no risk factors for aortic dissection (BAV, CoA and/or increased aortic dimensions) should be re-evaluated every 5-10 years. In patients considered to be at high risk, CMR should be repeated sooner.21Table 4 summarizes a reasonable imaging approach for these patients.
In the acute setting, CMR is not useful due to the relatively long examination time and difficulty in monitoring unstable patients. ECG-gated CT aortography is a valuable alternative for obtaining images of the entire aorta and aortic branch vessels including coronary arteries.5
PregnancyAs mentioned above, ovarian failure is a typical feature in TS with no or incomplete development of secondary sexual characteristics and primary amenorrhea. The X chromosome locus responsible for primary ovarian insufficiency in TS is probably on the short arm (Xq). However, 2-8% experience spontaneous pregnancy, and advances in assisted reproductive medicine with donated oocytes have been used, enabling successful pregnancies to be achieved.3,56
These pregnancies carry substantial fetal and maternal risks, exerting significant stress on the heart and aorta (with an increase in stroke volume and rise in estrogen level) and hypertensive disorders such as pre-eclampsia that may directly promote vascular damage and aneurysm formation. If uncontrolled, this can lead to aortic rupture and dissection.3,18
The risk of death during pregnancy from aortic dissection and rupture may be 2% or higher in women with TS.57
Thus, careful follow-up, including echocardiography and CMR performed by trained specialists familiar with TS, is essential for patients who intend to become pregnant.58
According to the 2008 publication of American Society of Reproductive Medicine, TS is a relative contraindication for pregnancy, but an absolute contraindication if there is a documented cardiac anomaly. However, the French task force 2011 recommendations, summarized in Table 6, do not list an isolated BAV as a contraindication, but rather a risk factor.59,60
Contraindications to pregnancy in TS according to American Society of Reproductive Medicine. Adapted from Cabanes et al.59
| Contraindications to Pregnancy in Turner syndrome | |
|---|---|
| Cardiovascular contraindications | History of aortic surgery History of aortic dissection Aortic dilatation with the largest ASI >2.5 cm/m2 or absolute dimension>3.5 cm Aortic coarctation Uncontrolled hypertension despite treatment |
| Hepatic contraindications | Portal hypertension with esophageal varicose veins |
If the ASI is less than 2.5 cm/m2 or the aortic diameter is less than 3.5 cm, and if there is no associated CoA, pregnancy can be authorized although careful CV monitoring is required, with an echocardiography suggested at the end of the first and the second trimesters and monthly during the third. If a patient is pending oocyte donation and the aorta increases by 10% or more in the annual echocardiography examination, said increase must be confirmed via a second imaging technique (CMR, cardiac CT or transesophageal echocardiography) and, if confirmed, pregnancy is contraindicated.59
Final remarksIn TS, the detection of congenital CV anomalies and their continuous monitoring with the use of cardiac imaging methods, particularly CMR (now recommended for optimal cardiac risk assessment), is of vital importance.
Aortic dissection, an extremely common event in these women, can occur at a remarkably young age and in pregnancy. Therefore, special attention to prevention and treatment should be given by monitoring the standard CV risk factors in TS. This approach is crucial to avoid life-threatening complications.
Pregnancy, which is not often spontaneous and can be achieved through assisted reproduction, increases risk in women with TS. Contraindications to pregnancy are defined, but even if none are present, patients should be carefully monitored before and during pregnancy.
To sum up, it is clear that TS is associated with several CV anomalies and risk factors present at an early age, which contribute to high TS mortality and morbidity. However, there are numerous unresolved issues that require further research.
Firstly, studies are warranted to clarify the pathophysiology of CV risk and the development of congenital CV anomalies. TS aortopathy has been postulated as a connective disorder, a theory that has not yet been confirmed. Determining the etiology will enable offering potential therapies to prevent aortic dissection. It will also provide information about potential risk stratification and, therefore, help determine the frequency of cardiovascular screening (which is currently not appropriately addressed).
Furthermore, the cut-off for aortic dilatation that puts an individual at high risk of aortic dissection in TS remains unclear. As such, the relationship between aortic dimensions and aortic dissection needs to be defined, and the adjustment of aortic dimensions for younger patients and those with lower BSA needs clarification.
Finally, the predictive values of aortic dimensions for prophylactic aortic surgery to prevent aortic dissection are undefined, as are the efficacy and safety of the procedure.
Conflicts of interestThe authors have no conflicts of interest to declare.
