



Due to the success of finding treatments for various types of cancer, overall cancer survival has increased substantially in recent decades. Ironically, the clinical success of antineoplastic therapy is attenuated by the comorbidity of cardiovascular diseases, which appear to be the main complications of intense cancer treatment and are the second main cause of long-term morbidity and mortality among cancer survivors.
Cardio-oncology is the new area of cardiology that aims to treat the specific cardiologic status of cancer patients.
Most antineoplastic therapies are associated with some degree of cardiovascular toxicity, ranging from asymptomatic and transient cardiac events to clinically significant and long-lasting events.
Antineoplastic agents are responsible for different cardiovascular injuries, which can be reversible or permanent. All anatomical structures of the heart can, however, be affected by transthoracic radiotherapy.
Cardiotoxicity mechanisms are recognized according to the presence or absence of structural anomalies and their reversibility, being classified into Type I and Type II, aiming to distinguish the drugs that induce irreversible damage (Type I) from the drugs that predominantly induce reversible left ventricular dysfunction (Type II).
While anthracyclines and anti-HER2 agents form the two major groups of cardiotoxic drugs, other antineoplastic agents, such as other monoclonal antibodies, certain tyrosine kinase inhibitors and antiangiogenic drugs can also be cardiotoxic via different mechanisms.
This article presents the different pathophysiological mechanisms of cardiovascular toxicity according to the treatment regimen used.
Devido ao sucesso obtido em encontrar tratamentos para vários tipos de cancro, a taxa de sobrevida global do cancro aumentou substancialmente nas últimas décadas. Ironicamente, o sucesso clínico da terapêutica antineoplásica é atenuado pela comorbilidade das doenças cardiovasculares, que surgem como as principais complicações do tratamento oncológico intenso e constituem a segunda principal causa de morbilidade e mortalidade a longo prazo entre os sobreviventes de cancro.
A cardio-oncologia é a nova área da cardiologia que visa tratar as situações cardiológicas dos doentes oncológicos com toda a sua especificidade.
A maioria das terapêuticas antineoplásicas está associada a algum grau de toxicidade cardiovascular, variando desde eventos cardíacos assintomáticos e/ou transitórios a eventos clinicamente significativos e de longa duração.
Os agentes antineoplásicos são responsáveis por diferentes lesões cardiovasculares, que podem ser reversíveis ou permanentes. Por outro lado, todas as estruturas anatómicas do coração podem ser afetadas pela radioterapia transtorácica.
Os mecanismos de cardiotoxicidade são reconhecidos, de acordo com a presença ou não de anomalias estruturais e sua reversibilidade, classificando-se em Tipo I e Tipo II, procurando distinguir os fármacos que induzem danos irreversíveis (Tipo I) dos fármacos que induzem predominantemente disfunção ventricular esquerda reversível (Tipo II).
Enquanto antraciclinas e agentes anti-HER2 formam os dois grandes grupos de fármacos cardiotóxicos, outros agentes antineoplásicos, como outros anticorpos monoclonais, certos inibidores de tirosina-kinase e fármacos antiangiogénicos, também podem ser cardiotóxicos através de diferentes mecanismos.
Neste artigo apresentam-se os diferentes mecanismos fisiopatológicos de toxicidade cardiovascular de acordo com o esquema de tratamento usado.
Cardiovascular diseases and cancer are the most prevalent diseases in the developed world,1 and the two leading causes of morbidity and mortality globally, accounting for 70% of medical causes of death worldwide.2
Regarding Portugal, the 2020 edition of “Health Statistics 2018”, published by the National Institute of Statistics, shows that circulatory system diseases and malignant tumors continued to be the two main basic causes of death in Portugal in 2018, in very similar proportions to the previous year. Together, and as in 2017, these two groups of diseases were responsible for about 54% of deaths in the country.3
In 2018, people in the country died mainly from circulatory system diseases (32 926 deaths) representing 29.0% of all deaths. Within the group of causes influenced by circulatory system diseases, 11 235 deaths were due to cerebrovascular diseases and 7241 were due to ischemic heart disease.3
In 2018, malignant tumors were the second leading underlying cause of death in the country with 28 531 deaths accounting for 24.6% of all deaths.3
Shared risk factors and the integrated prevention strategyCardiovascular disease (CVD) and cancer share common risk factors such as obesity, sedentary lifestyle, arterial hypertension (AHT), smoking, diabetes mellitus, older age group and dietary habits.4 Therefore, the co-prevalence of cancer and CVD is high.4 Diagnosis and prevention of both diseases should occur in an integrated manner.5
Indeed, from a public health perspective, there are several shared risk factors for CVD and cancer, and in addition to the aforementioned traditional risk factors, these now include pollution, consumption of processed meats, and new threats such as the issue of vaping, which is gaining popularity among younger people at an alarming rate. While these risk factors can be addressed at an individual level, a combat strategy targeting the population's health as a whole will be important.6
There appear to be shared pathways in the development of CVD and cancer, the role of inflammation being studied in both diseases, which is not surprising considering the shared risk factors.6
The need for cardio-oncologyDespite these facts, it is true that over the past 30 years, cancer mortality has decreased significantly due to early detection strategies, and advances in surgical approaches and antineoplastic therapy.7–9
Due to the success in finding treatments for various types of cancer, the overall cancer survival rate has increased substantially in recent decades, totaling aproximately 14.5 million cancer survivors in the United States.10
Improved survival rates may, however, be associated with damage to other organs, including an impact on cardiovascular health.11 CVD is now the second leading cause of long-term morbidity and mortality among cancer survivors.7–9,12–14 Ironically, the clinical success of antineoplastic therapy is attenuated by the comorbidity of CVD, which emerges as the primary complication of intense cancer treatment.15
Conventional chemotherapy (CT), new antineoplastic drugs with different types of action, and transthoracic radiotherapy (RT) can result in different expressions of cardiovascular damage that may be permanent or reversible in nature.16
Physicians following oncologic patients should have an understanding of:
- •
the possible cardiotoxicity of the antineoplastic agent or transthoracic RT being used;
- •
cardiovascular risk factors or preexisting CVD, predisposing the development of cardiotoxicity;
- •
use of preventive and therapeutic measures during possible cardiotoxic therapy;
- •
the fact that complications of antineoplastic therapy may only manifest themselves years after treatment;
- •
the older population may suffer from both cancer and CVD at the same time, and should receive coordinated treatment by the two specialties;
- •
the increase in the incidence of cancer, but also in survival rates, with a subsequent increase in cardiovascular complications in survivors.
It is in this context that cardio-oncology has emerged. Cardio-oncology is the new area of cardiology that aims to treat cardiologic status of cancer patients specifically, with the purpose of preventing the cancer treated patients from suffering from heart failure (HF) or dying from cardiovascular causes secondary to the antineoplastic therapy.16
This review article aims to present the mechanisms of cardiovascular toxicities secondary to antineoplastic therapies.
Most antineoplastic therapies are associated with some degree of cardiovascular toxicity, ranging from asymptomatic and/or transient cardiac events to clinically significant, long-term events.1
Cardiovascular toxicity secondary to antineoplastic therapy in oncologic patients – general principles:Antineoplastic agents are responsible for different cardiovascular injuries, which may be reversible or permanent. All anatomical structures of the heart can be affected by transthoracic RT.16
Cancer patients receive multiple antineoplastic drugs, sometimes alongside radiation, potentially having a series of cardiotoxic effects resulting from interactions between the different therapeutic modalities.17
Anthracyclines are the cornerstone of chemotherapy. These are direct cellular toxicants and are among the most powerful antineoplastic drugs, with well-known cardiac toxicity. More recently, with the introduction of signaling pathway inhibitors, such as human epidermal growth factor receptor 2 (HER2) signaling pathway inhibition by monoclonal antibodies (trastuzumab, pertuzumab, trastuzumab-emtansine) or tyrosine-kinase inhibitors (lapatinib), or vascular endothelial growth factor (VEGF) inhibitors, it has been found that these signaling inhibitors can also interact with cardiovascular signaling pathways and they can, therefore, have functional or structural effects on the myocardium leading to cardiovascular injury, especially if their use is associated with direct cellular toxic agents.18
The mechanisms of cardiotoxicity are recognized according to the presence or absence of structural abnormalities and their reversibility. In type I (model type: anthracyclines), cell apoptosis and myocardial necrosis occur in a dose-dependent manner, causing permanent damage (visible on biopsy), and early diagnosis is essential. In type II (model type: trastuzumab), cellular dysfunction occurs without apparent structural changes, due to the blockade of cell survival pathways associated with HER2. There seems to be no cumulative effect and the injury is reversible in most cases with discontinuation of the drug.18 It should be noted that this cardiotoxicity is potentiated by the association of anthracyclines with trastuzumab, in which case the expected reversibility may not occur with discontinuation of the drug or may only be partial.
This model, proposed by Ewer et al., to distinguish drugs that induce irreversible damage (Type I) from drugs that induce predominantly reversible left ventricular dysfunction (Type II), although beneficial, has limitations: for example, trastuzumab can trigger irreversible cardiac damage in patients with prior heart disease or potentiate anthracycline Type I cardiotoxicity.19
Signaling pathway inhibitors, such as monoclonal anti-HER2 antibodies and angiogenesis inhibitors, predominantly affect cardiac metabolism and contractile proteins, leading to transient contractile dysfunction.19 In most cases, when an echocardiographic diagnosis is made, there is a decrease in left ventricular ejection fraction (LVEF) in asymptomatic patients or in patients with a clinical manifestation of fatigue from moderate effort.
On the other hand, in the case of conventional CT agents such as anthracyclines, antimetabolites, and cyclophosphamide, there may be permanent myocardial cell damage by various mechanisms, and by cardiac remodeling.
While anthracyclines and anti-HER2 agents form the two major groups of cardiotoxic drugs, other antineoplastic agents such as other monoclonal antibodies, certain tyrosine-kinase inhibitors, and antiangiogenic drugs can also be cardiotoxic through different mechanisms.
Pathophysiologic mechanisms of cardiovascular toxicityType 1 toxicity agentsAnthracyclinesAnthracyclines (doxorubicin, epirubicin, daunorubicin, and idarubicin) are highly effective in the treatment of solid tumors and hematologic neoplasms and avoiding their use due to potential cardiac effects may negatively affect prognosis. Anthracyclines can, however, cause irreversible cardiac damage and thus negatively affect the patient's prognosis.17
Several mechanisms of cardiac cell injury by anthracyclines are proposed based on animal studies and cell culture systems, and it is not yet clear which ones are at work in the clinical setting.20
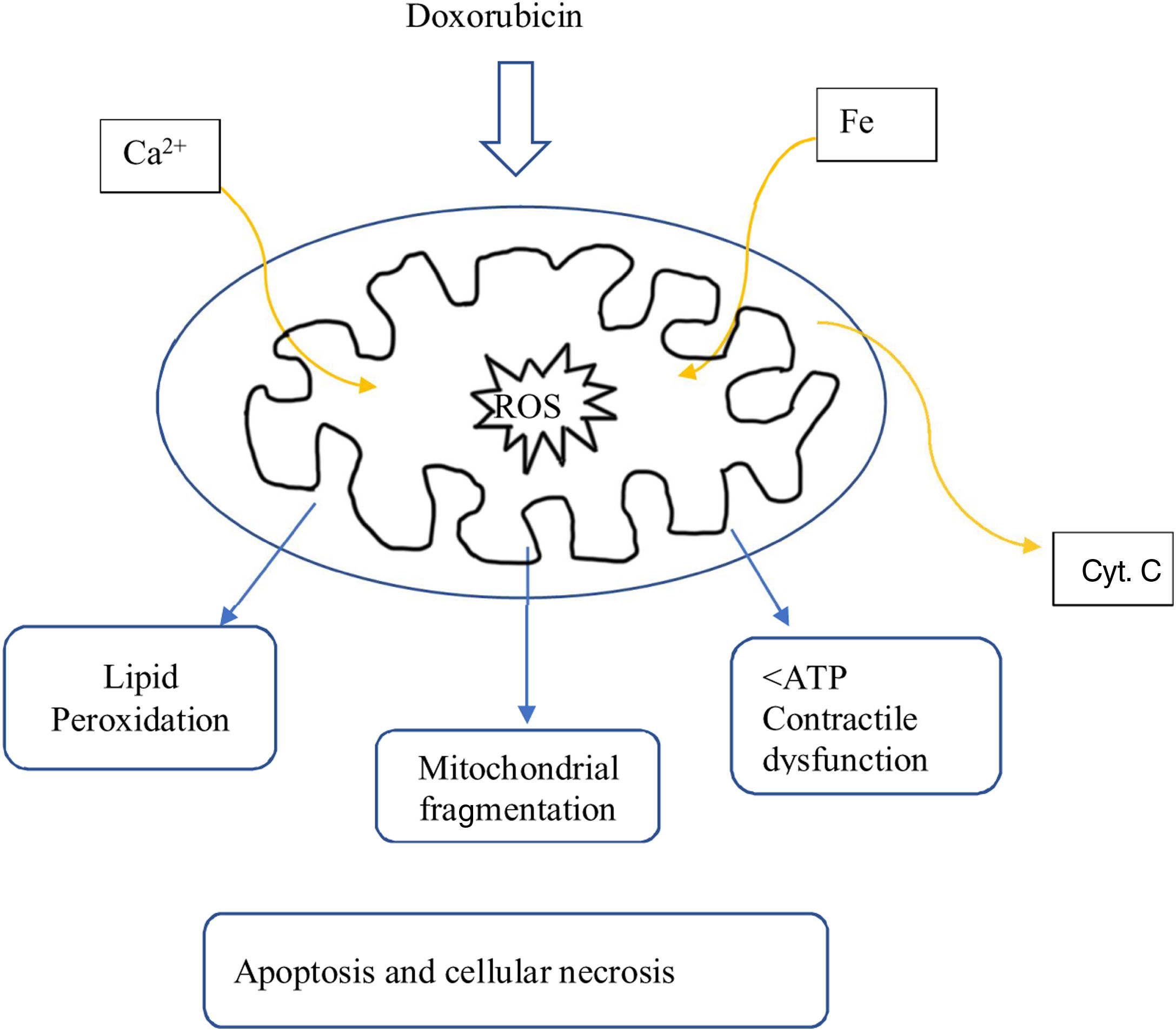
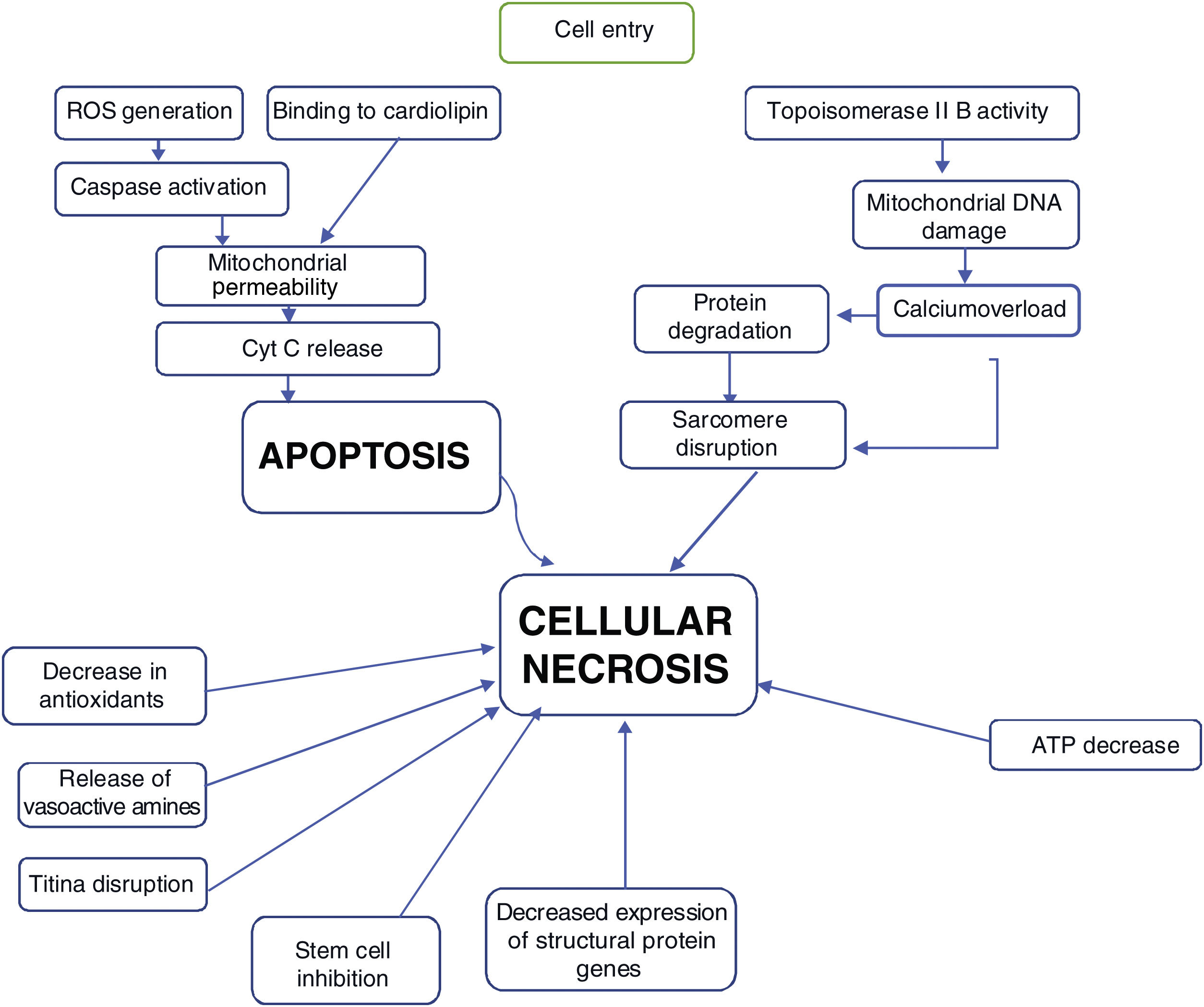
Most of the proposed mechanisms involve oxidative stress induced by anthracyclines, which suggests the generation of reactive oxygen species (ROS) and lipid peroxidation of the cell membrane are responsible for the injury to cardiomyocytes (Figure 1).17
However, there is considerable variability among patients in their susceptibility to anthracyclines. While many tolerate anthracyclines at standard doses without long-term complications, treatment-related cardiotoxicity may occur soon after the first dose in some patients.17
The mechanisms of doxorubicin's therapeutic effects on cancer cells include: generation of oxygen free radicals, to become interspersed with DNA, leading to inhibition of macromolecule synthesis, DNA binding, as well as DNA damage achieved by inhibition of topoisomerase IIB, and induction of apoptosis by inhibition of topoisomerase II.16
The first step in the process is the entry of the drug into the myocardial cell.
The proposed mechanism for the cardiotoxicity of doxorubicin is increased oxidative stress, demonstrated by increased levels of ROS and lipid peroxidation. Within the cell, the first mechanism is the formation of iron complexes and the production of oxygen free radicals that cause impaired mitochondrial function (Figure 1).16
Several mitochondrial enzymes such as NADH dehydrogenase, cytochrome P-450 reductase, and xanthine oxidase are involved in the generation of oxygen free radicals. Doxorubicin also increases superoxide formation by activating endothelial nitric oxide (NO) synthase.16
In addition to the formation of oxygen free radicals induced by the quinone portion of anthracyclines, oxidative stress that occurs via induction of NO synthase, forms NO and peroxynitrite, and this mechanism is associated with nitration and inactivation of key enzymes in the heart, namely myofibrillar creatine kinase.20
The oxygen free radicals produced by doxorubicin metabolism in cardiomyocytes can subsequently cause cell death through apoptotic pathways initiated by activation of caspase-9 and caspase-3, which leads to the opening of the mitochondrial membrane by altering the permeability of the transition pores and subsequent release of cytochrome C into the cytosol (Figure 1).16
Anthracyclines also cause compromise in membrane assembly and binding and in mitochondrial creatine kinase enzyme activity.20
Another mechanism of mitochondrial injury from doxorubicin may be through injury to the genome of mitochondria by interfering with topoisomerase IIβ activity.16
In the heart, as in other tissues, anthracyclines intersperse with nucleic acids and prevent the synthesis of DNA, RNA, and proteins. Some transcription regulatory proteins that appear important in regulating specific cardiac genes are particularly susceptible to the action of anthracyclines. The impaired synthesis of myofilament proteins, associated with accelerated myofilament degradation in the presence of anthracyclines, leads to a negative balance of sarcomeric proteins: the so-called “cardiac sarcopenia”.20
Anthracyclines also induce alterations in adenylate cyclase and adrenergic function, as well as abnormalities in calcium ion utilization, all critical processes in the dynamic regulation of cardiac function.20
The pathway to cardiomyocyte apoptosis also seems to undergo changes in the balance between cytoprotective and cytotoxic pathways: immediately after doxorubicin exposure, there is evidence that there is an initial positive regulation of the expression of the antiapoptotic proteins, Bcl-XL and Bcl-2, followed by decreases in their expression. This suggests that increasing the expression of Bcl-XL or Bcl-2 could protect against doxorubicin-induced cardiotoxicity.16
Cardiomyocyte death is thus determined by the balance between cytotoxic pathways and cytoprotective pathways. Understanding these cytoprotective pathways may provide insights into new possibilities to decrease anthracycline toxicity.16
The various mechanisms seem to contribute to progressive loss of myofibrils and cell death of cardiomyocytes by apoptosis and by necrosis with clear cell loss, thus leading to “heart muscle loss” (Figure 2).
The main RISK FACTOR for developing HF is the cumulative dose of anthracyclines.17,21
It is estimated that more than half of the patients who have taken anthracyclines will develop cardiac changes in the subsequent six years and are five times more likely to have HF than patients not treated with these drugs.21
In addition to the cumulative dose, there are other risk factors for antracycline toxicity (Table 1).
Risk Factors for anthracycline toxicity (adapted from Zamorano et al, 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity).
| Cumulative dose |
| Female gender |
| Age (>65 years or <18 years old) |
| Renal insufficiency |
| Previous or concomitant thoracic RT |
| Concomitant chemotherapy with- alkylating or antimicrotubule agents- immunomodulatory or targeted therapies |
| Pre-existing conditions:- cardiopathies- hypertension- genetic factors |
Legend: RT: radiotherapy.
Fluoropyrimidines (5-Fluoruracil and capecitabine) are antimetabolites that inhibit thymidylate synthase, preventing the production of thymidine and uracil, with subsequent inhibition of DNA and RNA synthesis, stopping the growth and division of rapidly dividing cells such as tumor cells.16
Capecitabine is the prodrug of 5-Fluoruracil (5-FU) and is administered orally, while 5-FU is administered intravenously. 5-FU is the third most commonly used chemotherapeutic agent in solid malignancies across the world, and the second most common chemotherapeutic drug associated with cardiotoxicity, after anthracyclines.
5-FU cardiotoxicity, manifested as coronary vasospasm, can occur with a wide dose range and it is dependent on the schedule and the route of administration. In general, risk appears higher with infusional regimens as opposed to bolus regimens: with bolus regimens, incidence of cardiotoxicity is lower.
There are predisposing factors for coronary vasospasm by fluoropyrimidines:
- •
pre-existing coronary heart disease;
- •
genetic deficit of dihydropyrimidine dehydrogenase;
- •
folic acid supplementation.
Histological lesions found include classic ischemic lesions and direct cytotoxic lesions.
The mechanism of toxicity is related to induction of endothelial injury with reduction of NO synthase and subsequent coronary vasospasm and cytotoxic injury of the endothelium with possible induction of thrombosis.16 The mechanism of coronary vasospasm is completely reversible. The clinical expressions of these lesions are:
- •
typical angina;
- •
acute myocardial infarction;
- •
arrhythmias, including ventricular tachycardia secondary to ischemia,
The incidence of myocardial ischemia varies considerably and can be as high as 10%, depending on the dose, schedule, and administration route.17
Electrocardiographic monitoring is essential for both the prevention and immediate diagnosis of toxicity. Once it has appeared, immediate discontinuation and replacement with another CT agent is indicated whenever possible.
While symptoms persist, treatment involves administering calcium channel antagonists such as verapamil and nitrates that are active against coronary spasm.16
Alkylating agentsAlkylating agents (cyclophosphamide, ifosfamide, busulfan, and other nitrogen mustards) are capable of alkylating different molecules (proteins, RNA, DNA) through an alkylating reaction with their alkyl groups.16 Cyclophosphamide and nitrogen mustards undergo complex modifications in the body that are responsible for their activation and inactivation and with consequent therapeutic and toxic activity. Alkylating agents differ in chemical structure, pharmacology, specific action, and clinical use (mainly in hematology and central nervous system cancer).
Toxicity is dose related, with the total dose of a single therapeutic cycle being more important than the cumulative dose. Concomitant use of anthracycline and RT increases the risk of cardiac toxicity.16
Heart failure has appeared in 7% to 28% of patients treated with cyclophosphamide, and there are a wide range of clinical manifestations of cardiotoxicity with this agent, varying from asymptomatic pericardial effusions to myopericarditis and HF.22
The anatomopathological features of alkylating agent toxicity consist of a framework of hemorrhagic myocarditis, edema, and cellular necrosis, with specific changes evident in the small intramural structures of the coronary arteries. The origin of these changes is an endothelial injury with microemboli and intracapillary lesions.16
Anti-microtubule agentsVinca alkaloids (vincristine, vinblastine) and taxanes (paclitaxel, docetaxel) are the two main groups of anti-microtubule agents, but their mechanisms of action are completely different: vinca alkaloids prevent microtubule formation, while taxanes prevent microtubule disassembly. In both, the cell cycle stops, leading to apoptosis.16
Paclitaxel can induce bradyarrhythmias and conduction defects directly through actions on the Purkinje system or indirectly through its transport vehicle. When used in combination with doxorubicin, paclitaxel appears to increase the cardiotoxicity of doxorubicin by altering its pharmacokinetics and increasing the formation of its major metabolite.16 Paclitaxel causes sinus bradycardia, auriculo-ventricular block, ventricular extrasystole, and ventricular tachycardia. Vinca alkaloids can cause angina, changes in the electrocardiogram, myocardial ischemia, and acute myocardial infarction.16
Cardiac events related to vinca alkaloids are more likely to occur in women than in men.16
The clinical presentation of Prinzmetal's angina and the reversible electrocardiogram sometimes described have led to the hypothesis of coronary spasm-induced ischemia.16
There is an increased risk of ischemic complications in patients with coronary artery disease or who have undergone previous thoracic RT.
PlatinumPlatinum compounds, when given in high doses, can cause HF and pericarditis.16 Higher plasma levels of cisplatin and carboplatin may result in decreased creatinine clearance and potentially lead to increased toxicity.16
Nephrotoxicity, seen in up to 35% of patients receiving cisplatin, can cause hypomagnesemia and hypokalemia, increasing the risk of cardiac arrhythmias.16
Patients of advanced age, after previous treatment with anthracyclines or with previous mediastinal irradiation, are at greater risk of cardiac toxicity from platinum.16
Type 2 toxicity agentsMonoclonal antibodies and tyrosine kinase inhibitorsMonoclonal antibodies are directed against cancer cell antigens such as CD52, VEGF, HER, CD20, HER2 and BCR-ABL tyrosine kinase (TK). A common type of toxicity is caused by a huge release of cytokines that can also lead to anaphylactic shock. The main class effects appear to be hypertension (HT) and heart failure (HF).16
The precise mechanism involved in monoclonal antibody-induced cardiac dysfunction is not fully understood but is completely different from that of anthracyclines and may be secondary to a sequential mechanism of cellular stress (Figure 3).
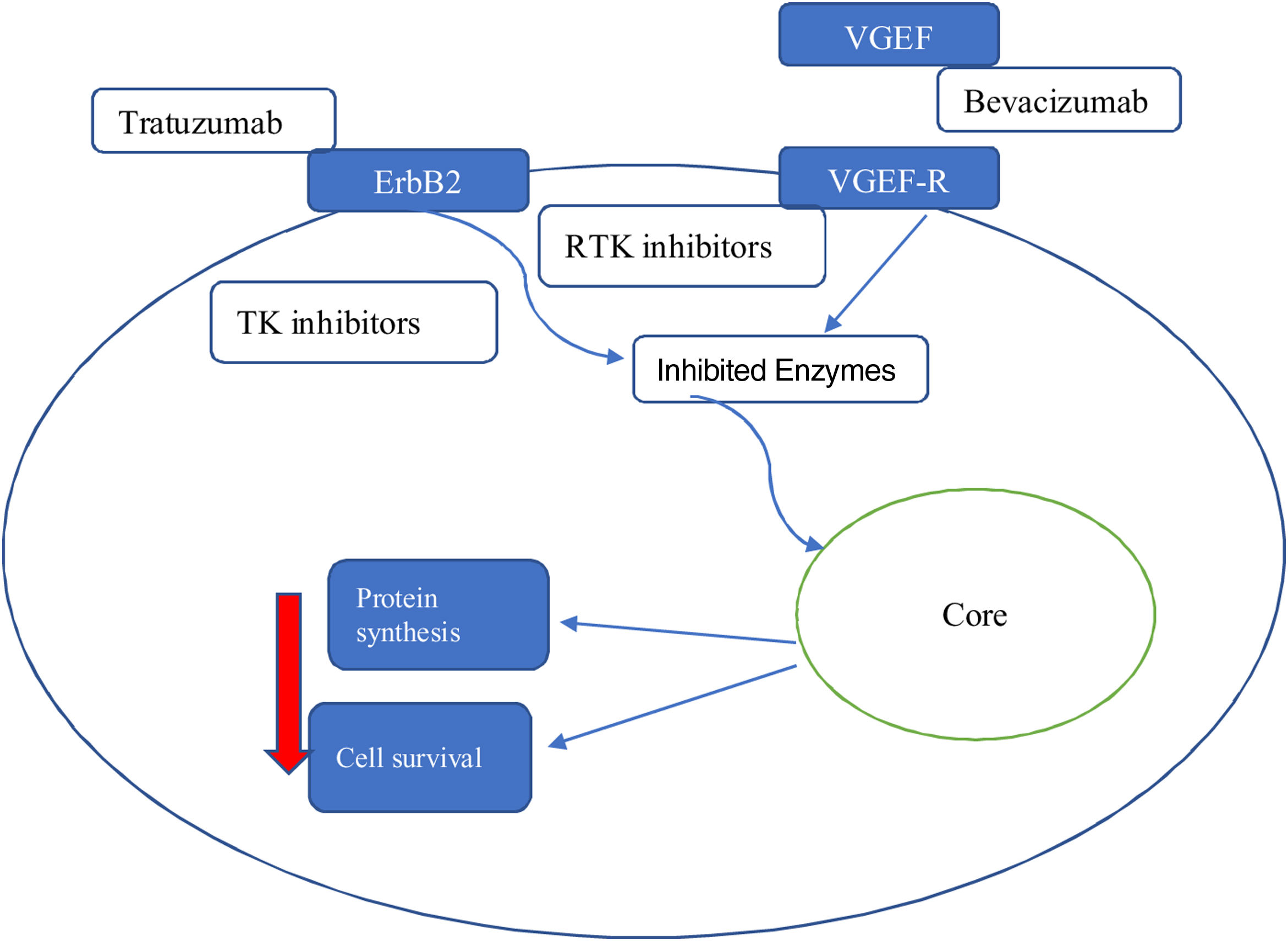
Since the approval of trastuzumab, several agents have entered the therapeutic arsenal, including small molecule TK inhibitors. It is difficult to make broad generalizations about these agents, because they generally have different targets within kinase enzymes.23
Tyrosine kinase enzymes are the key that allow for modulation of a variety of signaling pathways involved in the regulation of many important cellular functions. TK inhibitors act on specific pathways in neoplastic tissues that are influenced by the positive regulation of kinase enzymes.
Each molecule has, however, a specific toxicity profile due to the blocked receptors. It seems that the most problematic are agents that target VEGF and VEGF receptors. These agents are usually associated with severe systemic arterial disease, HT and ischemic events. The development of LVSD in these patients may be related to transient contraction impairment or increased afterload. Of most concern in this group are the non-selective agents, including sunitinib and sorafenib, because these drugs can affect up to 50 different kinases in addition to the target. Because these off-target kinases can play important roles in the heart and vasculature, the risk of toxicity is significant. As a result of its non-specific nature, it is difficult to give general recommendations on how to monitor patients receiving these agents.24
On the other hand, each of these drugs can have specific toxicity according to its specific cellular target.
Thus, it can be seen that:
- -
VEGF inhibitors can lead to vascular rarefaction and damage to NO production, which in turn leads to HT;
- -
in the case of HER2 pathway inhibitors, it is precisely the inhibition of this pathway in cardiomyocytes that seems to be responsible for the cardiotoxicity that appears with these drugs, such as trastuzumab and pertuzumab. In fact, the HER2 signaling pathway has an important protective, growth-promoting and antiapoptotic function at the cardiomyocyte level and is inhibited by these drugs. This toxicity is particularly aggravated by prior treatment with anthracyclines: the oxidative stress induced by anthracyclines leads to cell necrosis, but also to positive regulation of compensatory mechanisms by the HER2 pathway. If this is inhibited by monoclonal antibodies (mainly trastuzumab), the possibility of recovery is greatly reduced.16 The administration time between anthracyclines and trastuzumab probably has a crucial role in this toxicity (Figure 4).16
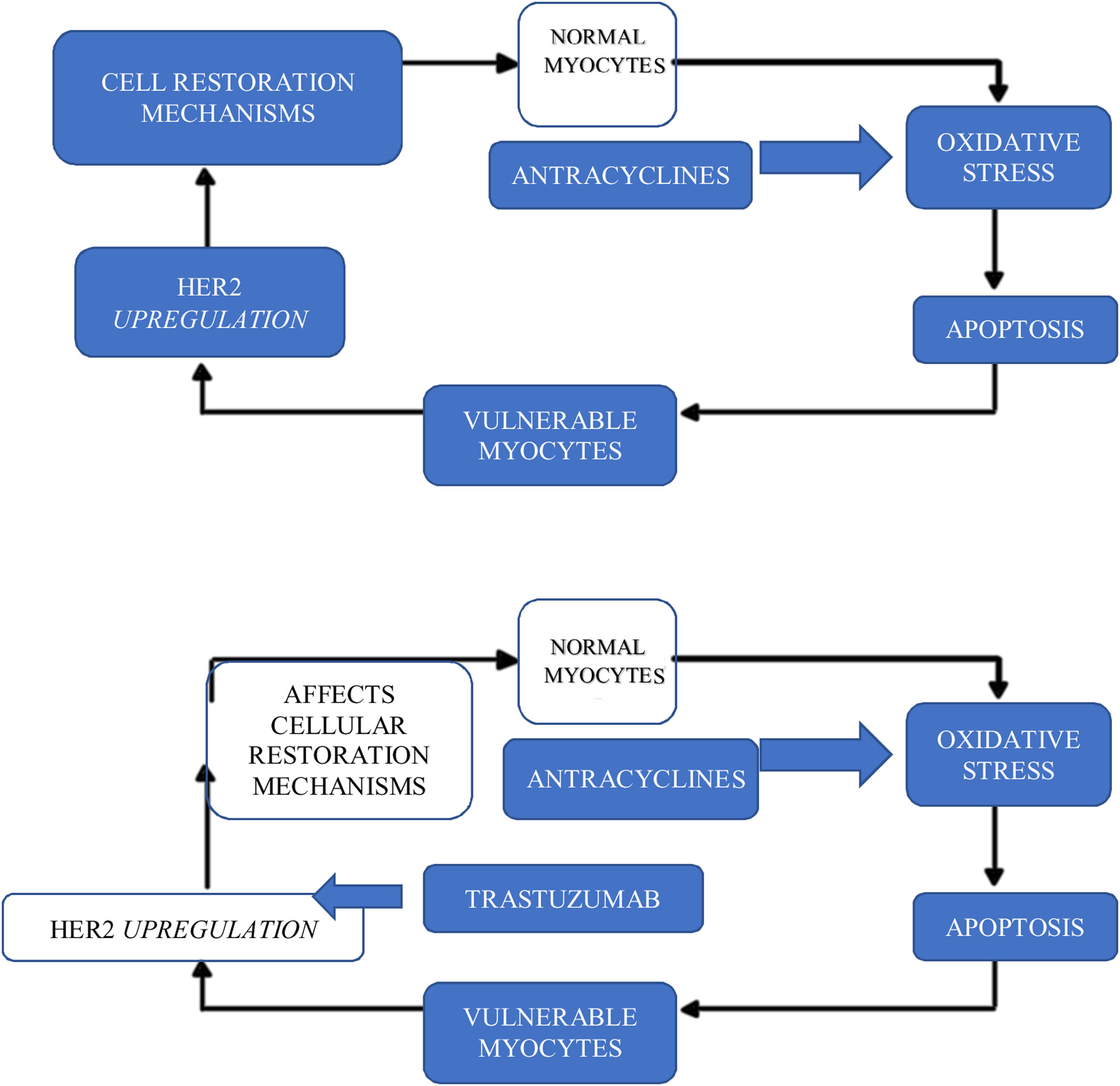
In these cases, it is clear that the most important risk factor for toxicity is prior administration of anthracyclines.
Inhibition of the Human Epidermal Growth Factor Receptor 2 pathwayHER 2 can be inhibited by any of the monoclonal antibodies - trastuzumab, pertuzumab or trastuzumab-emtansine (T-DM1) - or by TK inhibitors - such as lapatinib.
The data indicate that concomitant or prior use of anthracyclines substantially increases the cardiotoxicity of trastuzumab.
The cumulative incidence of cardiac dysfunction or HF in patients treated with anthracyclines and trastuzumab was 6.2% and 20.1% after 1 and 5 years, respectively.17
When trastuzumab was used concomitantly with antimetabolites and alkylating agents in patients with gastric neoplasm, the rates of dysfunction and HF were 5% and 1%, respectively.17
Unlike with anthracyclines, trastuzumab cardiotoxicity usually manifests during treatment. This has led to the implementation of different cardiotoxicity surveillance protocols.
Cardiotoxicity associated with trastuzumab is not thought to be dose cumulative, although the rate of LVSD in patients who were treated for 24 months was found to be twice that of patients treated for the usual 12 months.
Trastuzumab-induced LVSD and HF are usually reversible with cessation of trastuzumab and initiation of a cardioprotective strategy.
The mechanism of anti-HER2 drug-induced cardiotoxicity includes structural and functional changes in contractile proteins and mitochondria, but rarely leads to cell death, explaining the potential for reversibility.17
Risk factors for cardiotoxicity secondary to anti-HER2 drugs include:
- •
previous exposure to anthracyclines, with the time between anthracycline and anti-HER2 being of particular importance (from three weeks to three months);
- •
pre-existing HT;
- •
low LVEF;
- •
Older age.17
One of the most relevant clinical implications of trastuzumab-induced cardiotoxicity is that treatment interruption is associated with increased cancer recurrence.17
Although there are no randomized clinical trials to prove that HF drugs improve function in patients with trastuzumab-associated LVSD, by analogy with the experience of patients with anthracycline cardiotoxicity, the use of angiotensin-converting enzyme inhibitors as a cardioprotective strategy is advocated.
The risk of cardiotoxicity of other anti-HER2 therapies such as lapatinib, pertuzumab and T-DM appears to be similar to that of trastuzumab.17
Inhibition of the Vascular Endothelial Growth Factor pathway - antiangiogenic agentsVascular endothelial growth factor signaling pathway inhibitors, such as sorafenib and sunitinib, commonly cause HT. Although these are effective antineoplastic agents, their clinical use may be limited by their potential negative impact on cardiovascular health.14
HT is the most frequent form of cardiovascular toxicity observed with VEGF inhibitors, with a reported incidence of 19% to 47%.14 The mechanisms of hypertension induced by VEGF inhibitors have recently been reviewed and include:
- •
reduced NO production in the wall of arterioles;
- •
increased production of endothelin-1;
- •
capillary rarefaction that results in a reduction of effective capillary beds.14
In addition, induced hypertension may also be related to VEGF-mediated suppression of nephrin, a transmembrane protein important for glomerular slit diaphragm maintenance, which may contribute to proteinuria observed with this class of drugs.14 Strategies to prevent or control hypertension induced by inhibitors of the VEGF signaling pathway are critical to prevent cardiac dysfunction and early discontinuation of antineoplastic therapy.14
Currently, three antiangiogenic agents are widely used in oncology: bevacizumab, sorafenib and sunitinib. Each of the approved agents is associated with CVD.25
The antibody bevacizumab, which specifically binds to circulating VEGF, is associated with a 3% incidence of cardiac dysfunction.25 The two tyrosine kinase inhibitors, sorafenib and sunitinib, are less specific. At clinically relevant concentrations in in vitro kinase assays, sorafenib inhibits at least 15 kinases, including the VEGF receptor, and sunitinib inhibits more than 30 kinases, including the VEGF receptor.25
The rate of cardiotoxicity associated with sorafenib is still unclear; in one review, the rate of cardiotoxicity with sunitinib was estimated to be about 15%.25 Significant hypertension is seen with all three antiangiogenic agents.25
A 4% incidence of venous thrombo-embolism has been reported with sunitinib and sorafenib and a 12% incidence with bevacizumab.25
Bevacizumab may present with a more severe form of that, in some cases, does not seem to reverse with removal of the agent.25
Like tumors, the heart is highly dependent on adequate perfusion to perform its normal function, as well as VEGF-dependent pathways, just like the tumor. Studies show that VEGF inhibition causes microvessel rarefaction and reversible myocardial hibernation.25
Clinical manifestations of cardiovascular toxicity of antineoplastic therapyDespite the different mechanisms of toxicity, the cardiac response will usually go through one of the following presentations (Table 2):13
- a)
ventricular dysfunction and HF;
- b)
HT;
- c)
myocardial ischemia;d thromboembolism;
- d)
rhythm changes, conduction disturbances, or prolongation of the QTc interval.
Clinical manifestations of cardiovascular toxicity of antineoplastic therapy (adapted from TM Suter and MS Ewer).13
| Antineoplastic drug | Mechanism of toxicity | Cardiac response | Reversibility | Frequency |
|---|---|---|---|---|
| Anthracyclines | Apoptosis | Contractile dysfunction/HF | minimum | cumulative dosage |
| Cyclophosphamide | Myocarditis | Contractile dysfunction/HF | partial | rare |
| Cisplatin | unknown | Contractile dysfunction/HF | unknown | rare |
| Trastuzumab | Contractile protein dysfunction | Contractile dysfunction/HF | high | variable |
| Sunitinib | Mitochondrial dysfunction | Contractile dysfunction/HF | partial | low |
| Sorafenib | Contractile dysfunction/HF | unknown | rare | |
| Imatinib | Mitochondrial dysfunction | Contractile dysfunction/HF | high | rare |
| Anti-angiogenic (in general) | Endothelial dysfunction | hypertension | unknown | moderate dose-dependent |
| Pyrimidines | Direct Vasospasm | Myocardial ischemia | high | moderate |
| Cisplatin | Endothelial dysfunction | Thromboembolism | variable | moderate |
| Anti-angiogenic (in general) | Endothelial dysfunction | Thromboembolism | variable | moderate |
| Arsenic trioxide | Blockade of K+ channels | QT prolongation/arrhythmia | high | rare |
| Sunitinib | Blockade of K+ channels | QT prolongation/arrhythmia | unknown | rare |
Legend: HF: heart failure.
RT of thoracic tumors exposes the heart to radiation-induced complications. The pathophysiological and clinical consequences of cardiac irradiation have been studied mainly in patients with Hodgkin's lymphoma and breast cancer. The main cause of cardiac morbidity is radiation-induced coronary heart disease, with an estimated relative risk of between 2 and 3 according to studies.26
All anatomical structures of the heart can be affected by transthoracic RT.16 The aim is to reduce RT-induced cardiotoxicity by applying specific techniques that allow reduction of the exposed cardiac volume and the reduction of the radiation dose that reaches the heart. This work has been progressively developed by radio-oncology.
There are risk factors for cardiovascular toxicity from radiotherapy:16
- 1)
cumulative radiation dose (>30-35 Gy);
- 2)
dose per fraction (>2 Gy);
- 3)
irradiated heart volume;
- 4)
young age at the time of exposure;
- 5)
time passed since exposure;
- 6)
previous or concomitant chemotherapy (CT);
- 7)
traditional cardiovascular risk factors (hypertension, diabetes mellitus, dyslipidemia, and smoking).
This toxicity appears much less significant if the cardiac volume has received less than 30 Gy.26 The prolonged life expectancy of cancer patients and the increasing use of new cardiotoxic anticancer drugs underscore the ongoing need to further reduce the dose administered to the heart.26 In fact, 1 Gy added to the average cardiac dose would increase the cardiotoxic risk by 4% (95% confidence interval 2-6%, p=0.0002).26 The targets of RT toxicity are all cardiac structures, with the pathophysiological mechanisms being through acute inflammation of the small and medium arteries, endothelial injury, necrosis and16:
- 1)
diffuse interstitial fibrosis at the myocardial level - with subsequent diastolic dysfunction;
- 2)
fibrosis of the conduction system - with subsequent conduction defects, including second or third degree auriculo-ventricular blocks, which may require a pacemaker;
- 3)
pericarditis, pericardial effusion and pericardial fibrosis;
- 4)
faster formation of atherosclerotic plaques as well as rupture of these, especially in the proximal segments of the coronary arteries, manifesting as silent myocardial ischemia and acute coronary syndromes (often without previous symptoms);
- 5)
fibrocalcific degeneration of the left-sided valves, probably due to direct injury to the superficial endothelium of the valve itself, with evolution to aortic or mitral valve stenosis or regurgitation. This situation appears about 10 years after RT.
In the follow-up of patients who have previously undergone RT, it will be important to have:
- 1)
proper monitoring of known cardiovascular risk factors;
- 2)
screening for CVD, which usually starts 5-10 years after treatment.
Enhanced collaboration between the radio-oncologist and the cardiologist aims to detect and treat long-term complications following thoracic RT.26
ConclusionPrecision medicine in oncology requires an understanding of the mechanisms of cardiovascular toxicity in cardio-oncology.
It all starts at the molecular level and antineoplastic agents present specific toxicities according to their actions at the cardiomyocyte or endothelium cell level. On the other hand, in the irradiated heart in the field of thoracic RT, there is a phenomenon of pancarditis, evolving with the formation of fibrosis.
According to the recommended oncology treatment regimens, a certain cardiotoxicity profile can be expected. For this reason, cardio-oncology begins with a general cardiovascular approach, but then must act in a targeted manner according to the toxicity of each of the agents used.
For this clinical approach, it is important to understand the pathophysiological mechanisms of cardiovascular toxicity of the antineoplastic agents used, which was the focus of this article.
Conflicts of interestThe author has no conflicts of interest to declare.
