



Familial amyloid polyneuropathy type I (FAP type I) is a rare hereditary systemic amyloidosis caused by the Val30Met mutation in the transthyretin (TTR) gene. The clinical onset and spectrum are variable and depend on phenotypic heterogeneity. Cardiac complications (dysrhythmias and conduction disturbances, cardiomyopathy and dysautonomia) indicate a poor prognosis, even after liver transplantation. We report an atypical case of FAP type I, highlighting the severe cardiac involvement and its complications.
Early diagnosis of amyloid heart disease is increasingly important in the context of several clinical trials of promising new and experimental drugs.
A polineuropatia amiloidótica familiar tipo-I (PAF tipo-I) é um tipo raro de amiloidose hereditária e sistémica causada pela mutação Val30Met no gene da transtirretina (TTR). O início das manifestações e o espectro clínico podem ser variáveis e dependem da heterogeneidade fenotípica. As complicações cardíacas (disritmias e perturbações da condução, miocardiopatia e disautonomia) ditam um prognóstico mais reservado, mesmo após transplante hepático. É descrito um caso clínico atípico de PAF tipo-I com ênfase no grave envolvimento cardíaco e nas suas complicações.
O diagnóstico precoce de cardiopatia amiloidótica tem assumido relevância no âmbito de vários ensaios clínicos com fármacos experimentais promissores.
A 66-year-old white woman from Lisbon (Portugal) was being followed in the neurology department due to progressive sensorimotor and autonomic polyneuropathy of 12 years’ duration. Physical examination showed tetraparesis, thermal hypoesthesia and distal muscle atrophy of the lower limbs. Patellar and Achilles tendon reflexes were abolished. Electromyography revealed severe axonal sensorimotor polyneuropathy.
The most common diagnoses of polyneuropathy were excluded. A fat biopsy showed amyloid deposits. Genetic testing was performed and the Val30Met mutation in the transthyretin (TTR) gene was identified in heterozygosity.
The suspicion of amyloid cardiomyopathy had emerged at age 60, when the patient underwent transthoracic echocardiography (echo) during investigation of anginal pain. The echo findings suggestive of amyloid infiltration included an E/A-wave ratio of 0.6 (Doppler mitral inflow), left atrial (LA) dilatation (end-systolic diameter of 50 mm), mitral-aortic valvular thickening and a small pericardial effusion. Left ventricular (LV) dimensions were normal (50 mm in late diastole), with no hypertrophy and preserved systolic function.
One year later, in order to clarify episodes of syncope and dizziness, 24-hour ambulatory electrocardiographic (Holter) monitoring was performed. Supraventricular premature extrasystoles with compensatory pauses of up to 2.5 s were detected. Four months later, sinoatrial block (with some pauses longer than 4 s) was also identified on Holter.
Due to symptomatic bradyarrhythmia a dual-chamber pacing system (DDDR mode) was implanted, and the patient remained asymptomatic for the next five years.
At the age of 66, she was admitted to the cardiology department in heart failure (HF), NYHA class III. Physical examination revealed atrial fibrillation (AF) with a controlled ventricular rate and anasarca (total weight of 100 kg for a calculated ideal dry weight of about 60 kg). The echo examination showed LV wall thickening (15.11 mm), an enlarged LV cavity (58.47 mm at end-diastole), greater LA dilatation (56 mm) and diastolic transmitral flow with an almost restrictive pattern, with an E/A-wave ratio of 2.39 and deceleration time of 147 ms (Figure 1). Mild to moderate mitral regurgitation was also observed. Mild thickening of the aortic and mitral valves persisted as well as the small pericardial effusion, both findings observed six years previously.
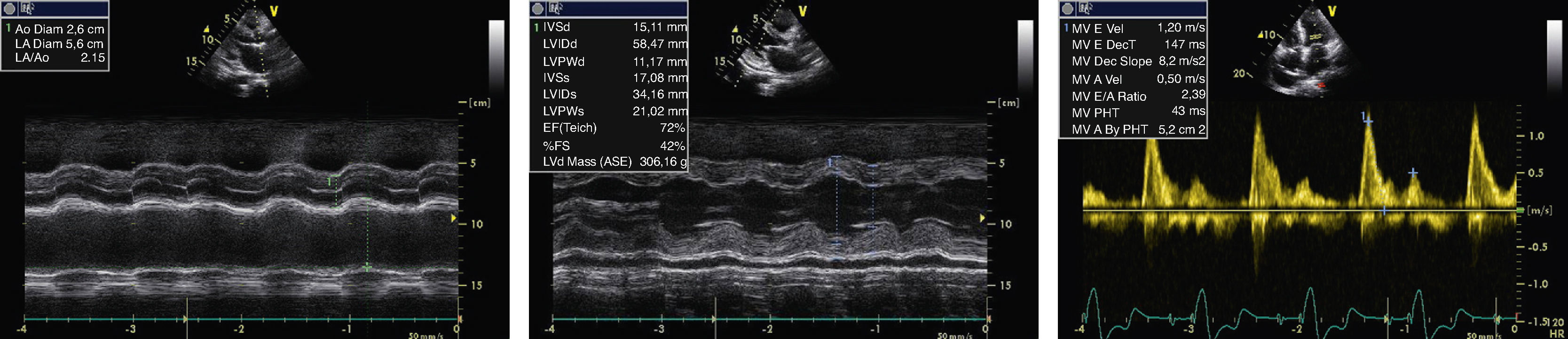
Echocardiographic findings at age 66. M-mode revealed a dilated left atrium (56 mm) (A), an enlarged left ventricular cavity (58.47 mm) with wall thickening (15.11 mm) and a small pericardial effusion (B). Doppler showed diastolic transmitral flow with an E/A-wave ratio of 2.39 and a deceleration time of 147 ms (C).
The patient's anasarca (also associated with nephrotic proteinuria and hypoalbuminemia) improved slightly with intravenous administration of albumin plus diuretics, but hemodialysis was needed. She lost 40 kg of body fluids and was transferred to the nephrology department.
The patient had mild and well-controlled systemic hypertension. There was no known family history of FAP type I. Both her parents had died of malignant disease at unknown ages. She has a daughter and a son, both healthy adults.
DiscussionThe term “amyloidosis” covers a heterogeneous group of rare diseases (acquired or inherited) characterized by extracellular deposition of abnormal insoluble fibrils resulting from protein misfolding.1–3 Accumulation of amyloid deposits in various tissues impairs their structure and function.1–3
Currently, there are 27 different types of amyloidosis classified according to their precursor protein.1 They may also be classified as systemic or local based on organ involvement.4,5
A definitive diagnosis of amyloidosis is usually made by biopsy specimen, in which Congo red staining reveals a typical red color under microscopy and apple-green birefringence under polarized light.2,3,6 But determination of the amyloid type is only possible by immunohistochemical techniques.6
The heart is usually affected, as part of systemic involvement.7 The most common systemic amyloidoses with myocardial involvement are acquired monoclonal immunoglobulin light-chain amyloidosis and TTR-related forms.4,5 Under the broad designation of amyloidosis, there are several clinically distinct entities that also require different treatment.5 But the severity of cardiac involvement in all forms is an important prognostic factor and early diagnosis remains a challenge.2
FAP type I is a rare autosomal dominant systemic amyloidosis caused by the Val30Met mutation in the TTR gene.8–11 It was first described by Corindo de Andrade, a Portuguese neurologist, in 1952.11 Higher prevalences in endemic areas of Portugal, Japan and Sweden have been reported.9,10 TTR is a plasma protein synthesized mainly in the liver and to a lesser extent in the choroid plexus and ocular tissues.10,12,13 It acts as a carrier for thyroxine and retinol-binding protein.10,13
A wide phenotypic heterogeneity has been found in FAP type I and is influenced by several factors such as genetic status (homozygosity or heterozygosity and penetrance), geographic distribution, age of onset and clinical spectrum.9,12,14
In Portugal, early-onset cases, usually before the age of 40, are more common than late-onset cases, which are rare and tend to occur in non-endemic areas.12,14
Symptomatic disease usually evolves over 10–20 years.10 Rapidly progressive sensorimotor polyneuropathy, which may be associated with dysautonomia, usually characterizes the initial clinical picture.13,14 Other clinical manifestations of cardiac, renal, ocular and gastrointestinal involvement often appear later.13,14
Cardiac involvement is common in hereditary TTR amyloidosis, but its clinical expression and severity depend on the amyloidogenic TTR variant.15,16
FAP type I is mostly linked with dysrhythmias and cardiac conduction disturbances.15–19 Amyloid infiltration into the subendocardium is probably the pathophysiological mechanism.19,20 Moreover, dysautonomia may also contribute to these disturbances, leading to sudden cardiac death (SCD).18,19 Cardiac conduction system involvement may result in sinus node dysfunction and atrioventricular and intraventricular block.19–21 Sinoatrial and/or atrioventricular block associated with Stokes–Adams syndrome requires pacemaker implantation15,19,22 as soon as possible, in order to alleviate symptoms and possibly prevent SCD.17 Supraventricular dysrhythmias associated with interventricular septum (IVS) thickening and LA dilatation have been reported.15 In patients aged over 60 a relationship between ventricular late potentials, non-sustained ventricular dysrhythmias and increased LV wall thickening has also been described.22
Besides autonomic dysfunction,23–25 ventricular dyssynchrony appears to be another mechanism responsible for the reduced heart rate variability observed in FAP type I, as it correlates with shorter LV filling time and prolonged total isovolumic time, regardless of QRS width.24
There are some reports of patients suffering hemodynamic instability and severe hypotension during anesthesia, particularly with acetylcholine-like anesthetics, due to the effects of dysautonomia on self-regulatory blood pressure mechanisms.25–27 This may be related to a discrepancy between cardiac muscarinic receptors and cardiac beta1-receptor regulation. Circulating catecholamines produced by the adrenal medulla maintain beta1-receptor sensitivity, while upregulation of muscarinic receptors may be due to lack of cholinergic neurostimulation.25
Amyloid cardiomyopathy with HF is an uncommon presentation in FAP type I.15,18,21 A strong relationship between age of onset, IVS thickness and LA dimension has been observed.15 Cardiomyopathy with mild LV thickening (>12 mm but below the usual value considered diagnostic for LV hypertrophy) may appear prior to the onset of polyneuropathy.15
Electrocardiography (ECG) and echo have an important role in the diagnosis of amyloid heart disease. Only a third of FAP type I patients show low-voltage QRS complexes due to amyloid infiltration.28 Dysrhythmias and conduction disturbances are frequently identified. Amyloid cardiomyopathy may be demonstrated when there is IVS end-diastolic thickness >12 mm on echo, after excluding other more probable causes.29 Echo findings other than LV wall thickening (with a granular sparkling appearance of the IVS) are a non-enlarged LV cavity, diastolic dysfunction, atrial septal thickening, atrial dilatation, valvular thickening and small pericardial effusion.2,7,29 In contrast to other types, echo signs in FAP type I are subtle.28
Early diagnosis of cardiac amyloidosis is of great value since it determines therapy and prognosis. But ECG and echo alone or in combination have low diagnostic accuracy for early detection of cardiac involvement. Moreover, only combining the findings of the two techniques (low-voltage QRS complexes and IVS thickening >19.8 mm) enables a diagnosis of cardiac amyloidosis to be made with a sensitivity of 72% and specificity of 92%.28
Other echo techniques (such as tissue Doppler and strain),3099mTc-DPD scintigraphy29, 123I-MIBG scintigraphy31 and serum NT-proBNP assay26 have shown promising results in detecting early signs of cardiac amyloid infiltration before the development of clinical manifestations and even abnormal echo findings on standard examination.
Liver transplantation is established as a therapeutic option to prevent disease progression because it suppresses pathological TTR production.32–34 Nevertheless, development and/or worsening of dysrhythmias and cardiac conduction disturbances,32,35 cardiac dysautonomia33 and amyloid cardiomyopathy36 have been reported after liver transplantation. Constant deposition of wild-type TTR preferentially on pre-existing cardiac amyloid deposits after liver transplantation has been suggested as a plausible mechanism.34,37
Myocardial involvement in FAP type I thus carries a poor prognosis even after liver transplantation, and new treatments that offer better control of heart disease are therefore needed. Recently, the new drug tafamidis, which stabilizes the native homotetramer, preventing the formation of cytotoxic species, has been approved by the European Medical Association for treatment of stage 1 FAP type I.38 However, its benefit in cardiac amyloidosis is not yet known. Clinical trials of experimental drugs that block the amyloidogenic process in several ways, such as resveratrol and its analogs,39 carvedilol,40 doxycycline41 and ribozymes42 have shown promising results for future treatment of cardiac involvement in FAP type I.
The case reported here has some unusual aspects. Our patient had late onset FAP type I (the clinical picture emerged after the age of 50). This is unusual in Portugal, although similar cases outside the endemic areas have been reported. However, the lack of information on the patient's genealogy means that no conclusions can be drawn.
Although the patient had no known family history of FAP type I, genetic testing confirmed the presence of the Val30Met mutation in the TTR gene. Her parents died without undergoing genetic screening, so three hypotheses may be considered. First, given the condition's phenotypic variability, her parents may have died prior to the clinical onset of the disease. If this was the case, they may also have had late-onset disease. Second, the parents may also have been asymptomatic owing to gonadal mosaicism. And third, a de novo mutation may have occurred in our patient.
The patient had sinoatrial block and AF, which are frequent complications of cardiac involvement. Stokes–Adams syndrome related to sinoatrial block was treated successfully with pacemaker implantation. Nevertheless the severity of amyloid cardiomyopathy as denoted by echo findings – including significant LV wall thickening and changes over time in Doppler mitral inflow pattern – is not typical in this type of amyloidosis. The same is true for the clinical severity of HF, which seems to be associated with amyloid cardiomyopathy and significant diastolic dysfunction. The patient's anasarca may be explained by a maladaptive response to congestive low-output HF as well as by nephrotic syndrome associated with renal amyloid infiltration.
ConclusionsThe condition's phenotypic heterogeneity, as well as the absence of a known family history, should be considered when FAP type I is a diagnostic hypothesis. This helps to avoid late and inaccurate diagnosis and enables counseling of the family with regard to genetic screening.
Symptomatic improvement and prevention of possible SCD can be achieved in patients with bradyarrhythmias by early pacemaker implantation.
Advances in diagnostic techniques that will allow detection of early signs of amyloid heart disease, together with the development of new therapies, are urgently needed to reduce mortality related to cardiac events in FAP type I patients and to offer them a better quality of life.
Due to its multisystemic nature, patients with FAP type I should be referred to specialized centers capable of providing a multidisciplinary approach.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this investigation.
Confidentiality of dataThe authors declare that no patient data appear in this article.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Oliveira Santos M, Brito D. Doença cardíaca grave numa situação invulgar de polineuropatia amiloidótica familiar tipo-I 2012. http://dx.doi.org/.
