





Congenital long QT syndrome (LQTS) is a rare hereditary disease, with an incidence of 1 in 2000, characterized by prolonged ventricular repolarization and malignant ventricular tachyarrhythmias. We report the case of a 30-year-old woman, previously diagnosed with neurocardiogenic syncope, in whom LQTS was identified. The patient received an implantable cardioverter-defibrillator due to polymorphic ventricular tachycardia under beta-blocker therapy. Molecular genetic testing identified three mutations in heterozygosity in the KCNH2, KCNQ1 and SCN5A genes, which is a rare finding and is associated with worse prognosis.
A síndrome do QT longo congénito (SQTLC) é uma doença hereditária rara, com uma incidência de uma em cada 2000 pessoas, caracterizada por uma repolarização ventricular prolongada e por taquiarritmias ventriculares malignas.
Reportamos o caso de uma doente de 30 anos, com diagnóstico prévio de síncope neurocardiogénica, em que identificamos a SQTLC. A doente implantou cardioversor-desfibrilhador implantável por taquicardia ventricular polimórfica apesar da terapêutica com bloqueador beta.
Foram identificadas nesta doente três mutações em heterozigotia nos genes KCNH2, KCNQ1 e SCN5A, achado raro que lhe confere um pior prognóstico.
Congenital long QT syndrome (LQTS) is a rare hereditary disease, with an incidence of 1 in 2000, characterized by prolonged ventricular repolarization and malignant ventricular tachyarrhythmias, typically polymorphic ventricular tachycardia or torsades de pointes, which can result in syncope or sudden death.1–3 Arrhythmic events usually first appear around the age of 12.
Case reportA 30-year-old woman had been followed in pediatric cardiology consultations for recurrent syncope since infancy. She had initially been diagnosed with epilepsy but tilt testing showed a vasodepressive response. Midodrine was prescribed, but without clinical improvement. No electrocardiographic records are available from that time.
She was referred by her health center for cardiology consultation in our hospital due to continuing recurrent syncope; she reported episodes of presyncope and syncope, occasionally preceded by palpitations and sometimes triggered by standing.
There was no other relevant medical history; her family history included the sudden death of her mother at age 46, with a history of type 2 diabetes and anatomopathological evidence of chronic ischemic heart disease.
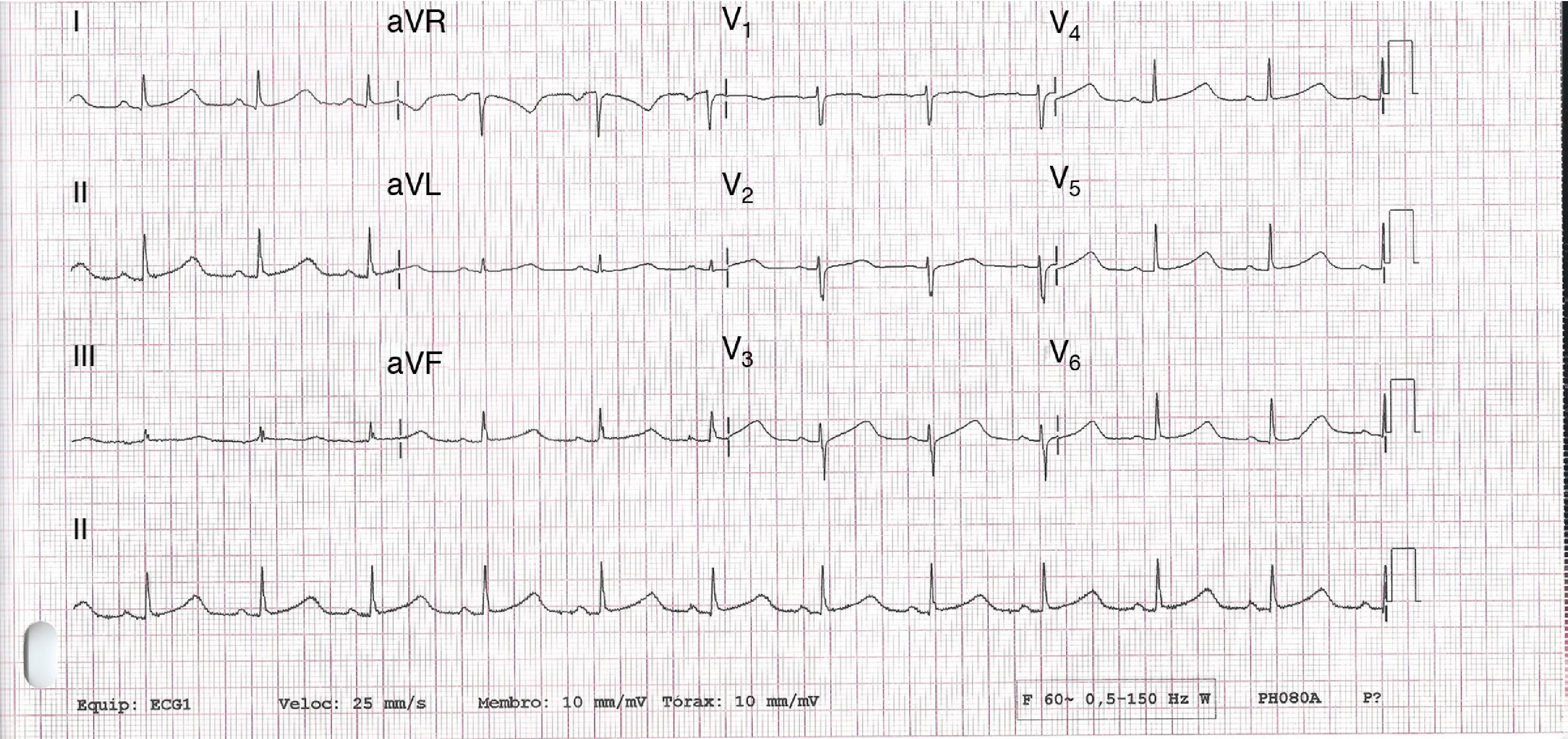
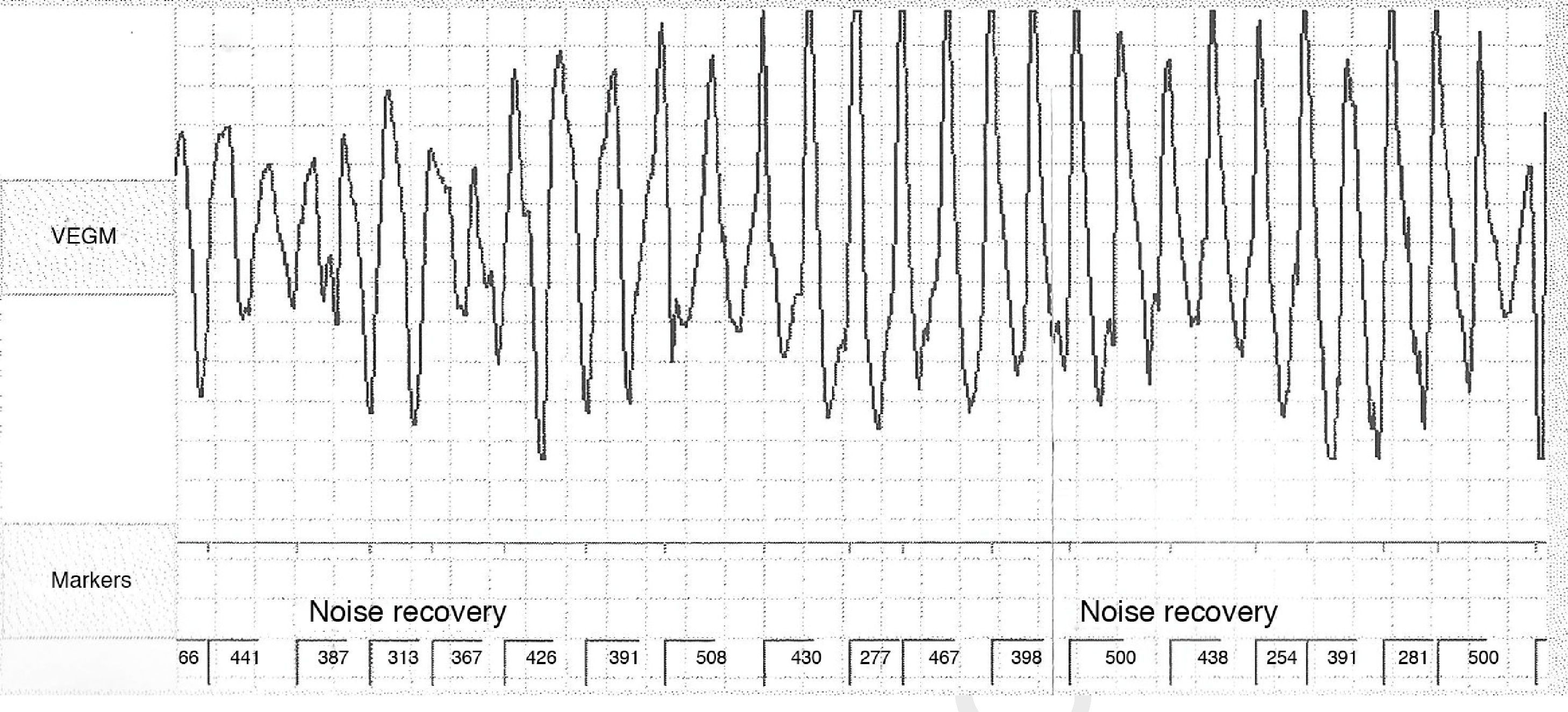
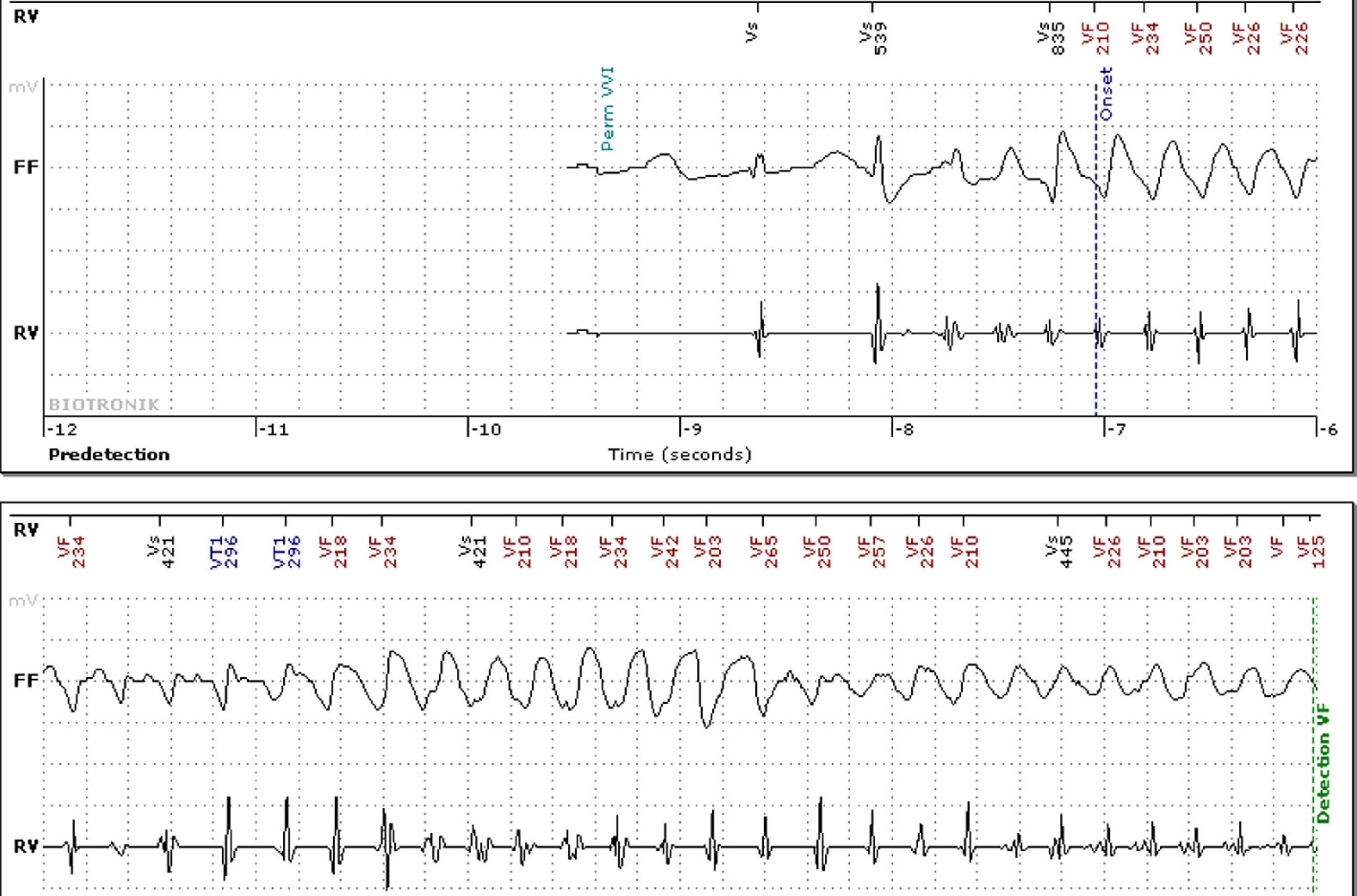
No abnormalities were detected on laboratory tests and transthoracic echocardiography revealed no significant alterations. The 12-lead electrocardiogram (ECG) performed at the cardiology consultation showed sinus rhythm and significant QTc prolongation (500–565 ms) (Figure 1). She was taking carvedilol 12.5 mg twice daily but the dose could not be titrated due to symptomatic hypotension. In view of the result of the previous tilt test and the non-specific symptoms, it was decided to implant an event recorder. The patient reported several episodes of pre-syncope, which corresponded to periods of sinus tachycardia, and in one of these nonsustained polymorphic ventricular tachycardia (VT) was recorded (Figure 2). It was accordingly decided to implant an implantable cardioverter-defibrillator (ICD). Around a month after the procedure remote monitoring recorded an appropriate shock for polymorphic VT (Figure 3). Her beta-blocker therapy was changed to propanolol.
Molecular genetic testing identified the c.785delG mutation in heterozygosity in exon 4 of the KCNH2 gene, which leads to the formation of a premature STOP codon in position 359 in the protein (p.Gly262AlafsX98). Two further mutations were found:
- –
c.535G>A, in heterozygosity, in exon 3 of the KCNQ1 gene, which leads to substitution of an amino acid at position 179 of the protein (p.Gly179Ser);
- –
c.3068G>A, in heterozygosity, in exon 17 of the SCN5A gene, which leads to substitution of an amino acid at position 1023 of the protein (p.Arg1023His).
LQTS is a genetic disease characterized by significant prolongation of the QT interval, predisposing to ventricular arrhythmias and manifesting clinically with syncope, seizures or sudden death in individuals with structurally normal hearts.1,2 The high incidence of vasovagal syncope and epilepsy in young individuals without structural heart disease means that diagnosis is often delayed or incorrect.4
According to the HRS/EHRA/APHRS expert consensus statement on inherited primary arrhythmia syndromes, LQTS is diagnosed: (a) in the presence of an LQTS risk score ≥3.5 in the absence of a secondary cause for QT prolongation and/or (b) in the presence of a pathogenic mutation in one of the LQTS genes or (c) in the presence of a QTc ≥500 ms in repeated 12-lead ECGs. LQTS can be diagnosed in the presence of a QTc between 480 and 499 ms in repeated ECGs in a patient with unexplained syncope in the absence of a secondary cause for QT prolongation or a pathogenic mutation.1
The Schwartz score estimates the probability of a diagnosis of LQTS on the basis of clinical, electrocardiographic and family history criteria. It has two main limitations: although its sensitivity is high its specificity is low; and it does not include genetic testing as a diagnostic criterion, failing to identify 30% of asymptomatic carriers.5–8
Mutations have been identified in several genes and new cases are frequently reported. The genes most often affected are KCNQ1 (LQT1), KCNH2 (LQT2) and SCN5A (LQT3).1–3,7,9 The first two lead to loss of function of potassium channels and the third causes gain of function in sodium channels. All three alter ion channels, destabilizing the membrane potential. Most mutations have an autosomal dominant inheritance pattern.
Clinical characteristics, particularly triggers of VT, and the electrocardiographic pattern of prolonged QT interval, can be used to correctly infer the genotype in 70–90% of cases.10 LQT1 is characterized by a broad T wave, and arrhythmias, being adrenergic-dependent, arise during exercise or emotional stress, while in LQT2 the T wave is narrow and notched and the most common triggers are emotional stress and auditory stimuli. In LQT3 the prolonged QT interval is mainly due to prolongation of the isoelectric ST segment, and events usually occur at rest or during sleep, demonstrating its low sensitivity to changes in the sympathetic nervous system and raising concerns regarding its response to beta-blocker therapy.7,10
It is important to exclude drug use, hypothyroidism and electrolyte imbalance as causes of a prolonged QT interval; although a long QT can arise as a result of pharmacological therapy, this may also unmask previously occult LQTS.
Genetic testing, which identifies the mutation responsible in 50–70% of cases, is important to confirm the diagnosis, identify asymptomatic carriers, stratify risk for arrhythmic events and provide a basis for genetic counseling.7 It is essential to identify silent carriers, since they are at high risk for severe ventricular arrhythmias. Those at greatest risk are patients with LQT1 and LQT2 with QTc >500 ms and male patients with LQT3 irrespective of QT interval. Digenic and compound mutations are found in 8% of patients with LQTS and are associated with a more severe phenotype.12,13
Our patient presented mutations in three different genes. The p.Gly262AlafsX98 mutation in the KCNH2 gene has been described in patients with LQTS (Human Gene Mutation Database [HGMD] CD070027) and is known to be a cause of the condition.14 The mutations p.Gly179Ser in KCNQ1 and p.Arg1023His in SCN5A have also been reported in LQTS (HGMD CM002321 and CM054857, respectively).14
In all patients with a clinical or molecular diagnosis, drugs that prolong QT interval are contraindicated, and any electrolyte disturbances should be identified and treated. Beta-blockers are recommended in asymptomatic patients with QTc ≥470 ms and symptomatic patients with syncope or ventricular arrhythmias (class I recommendation), and can be useful in asymptomatic patients with QTc <470 ms (class IIa). Sympathetic denervation is recommended in high-risk patients in whom ICD implantation is contraindicated or refused and for those in whom beta-blockers are either not effective in preventing arrhythmias or not tolerated (class I).1 ICD implantation is recommended in all patients who are survivors of a cardiac arrest and in those with VT or syncope under beta-blocker therapy.1,2 In patients with LQT3 and QTc >500 ms, sodium channel blockers can be useful in association with beta-blockers.1 There is evidence that nadolol and propanolol are the safest and most effective beta-blockers for these patients.11
ConclusionsLQTS is a rare hereditary disease in patients without structural heart disease that is associated with a high risk of malignant VT and sudden death. ICD implantation is indicated after resuscitated sudden death or persistence of symptoms despite beta-blocker therapy. The case presented is of a patient with LQTS in whom mutations were identified in three different genes, which is a rare finding and is associated with worse prognosis.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that no patient data appears in this article.
Right to privacy and informed consent.The authors declare that no patient data appears in this article.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Fernandes M, Martins Ribeiro S, Sanfins V, et al. Síndrome do QT longo: mutação trigénica, um caso raro. Rev Port Cardiol. 2015;34:359.
