





The authors present a case of systemic amyloidosis with cardiac involvement. We discuss the need for a high level of suspicion to establish a diagnosis, diagnostic techniques and treatment options. Our patient was a 78-year-old man with chronic renal disease and atrial fibrillation admitted with acute decompensated heart failure of unknown cause. The transthoracic echocardiogram revealed severely impaired left ventricular function with phenotypic overlap between hypertrophic and restrictive cardiomyopathy. After an extensive diagnostic workup, which included an abdominal fat pad biopsy, the final diagnosis was amyloidosis.
Os autores apresentam um caso de amiloidose sistémica com envolvimento cardíaco e discutem a importância de um elevado índice de suspeição para o diagnóstico, os meios de diagnóstico e as opções terapêuticas à luz do conhecimento atual. Homem de 78 anos, com antecedentes de doença renal crónica e fibrilhação auricular, admitido por insuficiência cardíaca aguda de etiologia desconhecida. O ecocardiograma transtorácico mostrou ventrículo esquerdo não dilatado, com compromisso severo da função sistólica global com overlap fenotípico de miocardiopatia hipertrófica e restritiva. Após estudo complementar alargado, o diagnóstico definitivo de amiloidose foi obtido por biópsia da gordura abdominal.
A 78-year-old man, with no cardiovascular risk factors but a history of paroxysmal atrial fibrillation and chronic renal disease (National Kidney Foundation stage IV), was admitted to the cardiac care unit for acute heart failure (HF) of unknown cause, in New York Heart Association (NYHA) class IV. He reported exertional dyspnea and worsening peripheral edema over the previous two months. He now complained of dyspnea on minimal exertion, orthopnea and paroxysmal nocturnal dyspnea, peripheral edema and increased abdominal circumference, apparently decreased urine output, and upper limb paresthesia. Physical examination showed anasarca, blood pressure 114/81 mmHg, heart rate (HR) 67 bpm and 90% oxygen saturation in room air.
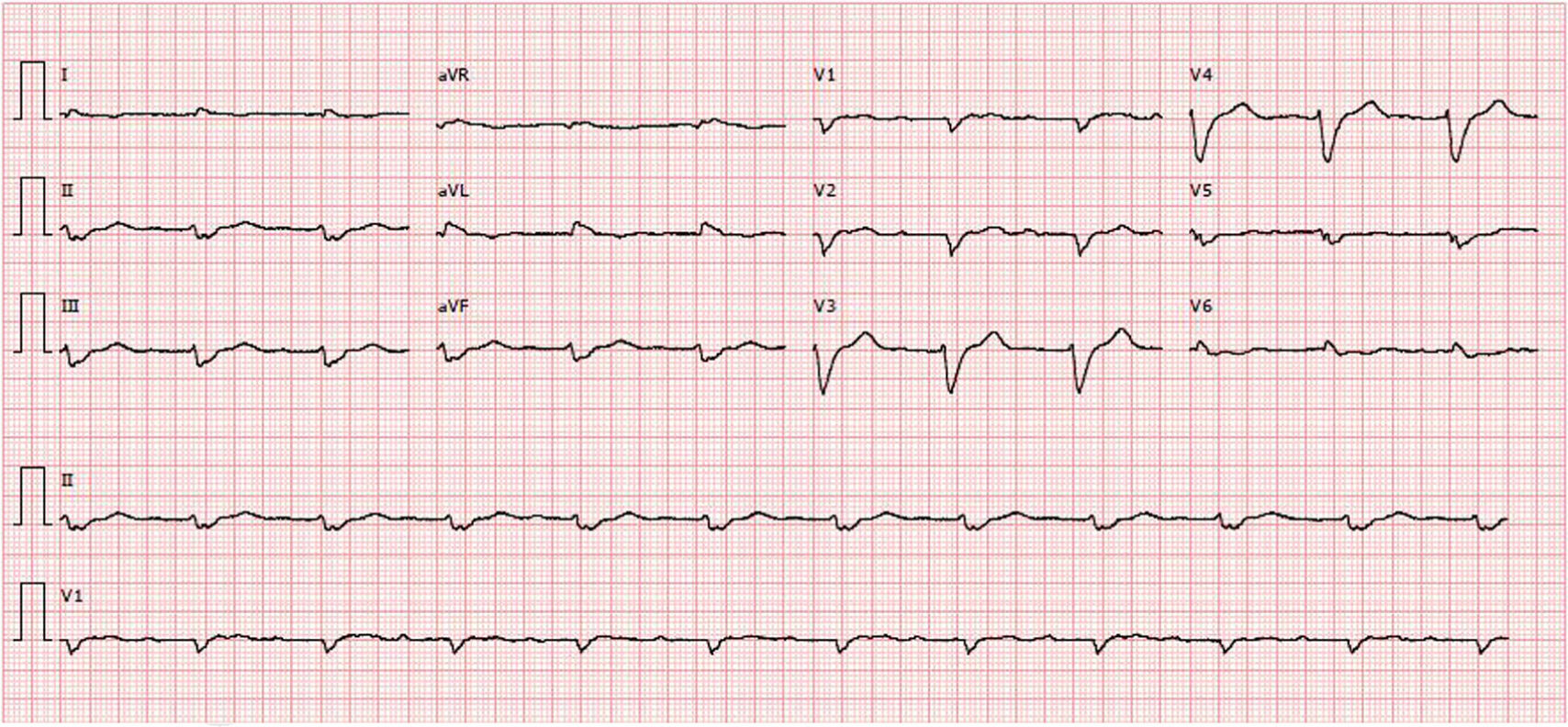
Diagnostic exams revealed type 2 respiratory failure with acidosis (pH 7.24; PaO2 58 mmHg, PaCO2 55 mmHg, HCO3 23.6 mmol/l, and lactates 2.66 mmol/l); normocytic anemia (hemoglobin 12.1 g/dl), with no increase in inflammatory markers; NT-pro-BNP 43300 pg/ml; hyponatremia (Na+ 132.7 mmol/l); worsening of chronic renal disease (urea 24.8 mmol/l and creatinine 207.8 μmol/l); and evidence of hepatic congestion (alkaline phosphatase 183 U/l), with increased transaminases (glutamic oxaloacetic transaminase 161 U/l and glutamic pyruvic transaminase 297 U/l). The chest X-ray showed alveolar interstitial infiltrate with a butterfly pattern. The electrocardiogram (ECG) showed sinus rhythm, HR 67 bpm, first-degree atrioventricular block, low voltage in the frontal leads and complete left bundle branch block (Figure 1).
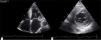
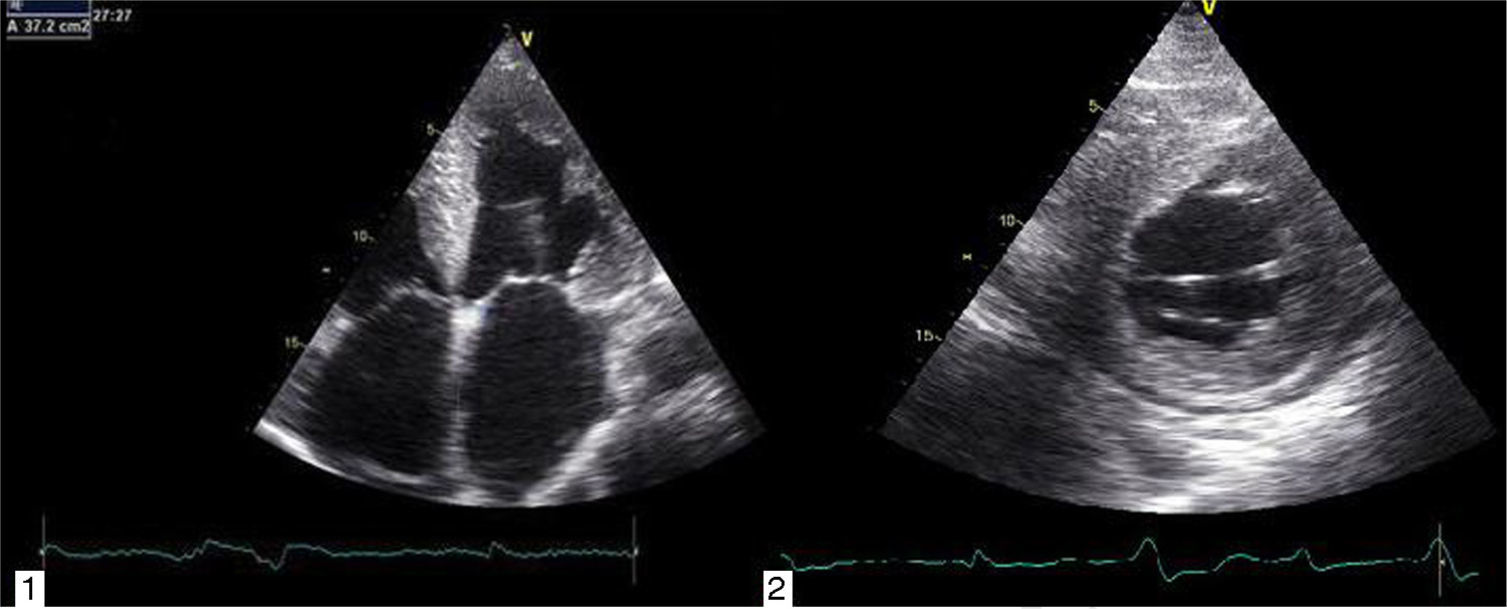
Transthoracic echocardiography revealed left ventricular (LV) size at the upper limit of normal (end-diastolic diameter 60 mm), with increased wall echogenicity, severe hypertrophy of the interventricular septum (20 mm) and moderate hypertrophy of the other walls (posterior wall 15 mm; LV mass index 200.1 g/m2); severe biatrial dilatation (left atrial area 37 cm2/m2) (Figure 2A and B); severely impaired LV systolic function (ejection fraction 27% by Simpson's method, with global longitudinal strain of −3%); hypocontractile right ventricle (tricuspid annular plane systolic excursion 12 mm); LV filling pattern suggestive of restrictive cardiomyopathy (deceleration time 145 ms, septal E′ 3 cm/s, lateral E′ 4 cm/s, and mean E/E′ ratio 29.5); right ventricular/right atrial gradient 43 mmHg; dilated inferior vena cava (2.3 cm) without inspiratory collapse; a small circumferential pericardial effusion; and interatrial septal hypertrophy.
Cardiac magnetic resonance imaging (CMRI) was not performed since the patient had a hip prosthesis.
Anticongestive therapy and a positive inotropic agent (levosimendan) were begun, together with noninvasive ventilation, resulting in progressive clinical improvement.
Given the findings of severe LV hypertrophy and low voltage on the ECG, a diagnosis of amyloidosis was considered. Abdominal fat pad biopsy confirmed the presence of amyloid deposits, exhibiting green birefringence after staining with Congo red.
Laboratory tests showed increased beta-2 microglobulin (4.98 mg/l), erythrocyte sedimentation rate 32 mm/h, C-reactive protein 5.0 mg/dl, calcium 2.07 mmol/l, phosphorus 1.05 mmol/l, parathyroid hormone 171 pg/ml, negative tumor markers and normal autoimmune parameters. Serum protein immunofixation revealed an IgG monoclonal spike (11.8 g/dl), with no changes in urine protein immunofixation. The ratio between kappa and lambda light chains was 1.22 (7.29:5.98 g/dl). After discussion with the hematology department, myelography and bone marrow biopsy were performed, which showed no plasma cell proliferation. No mutations were identified in genes coding for transthyretin proteins.
Based on the above findings, a diagnosis of systemic amyloidosis with cardiac involvement was made. The type of fibril involved was not determined, and 99mTc-DPD scintigraphy was not performed to establish the type of amyloidosis.
The patient was readmitted four months later for decompensated HF in NYHA class IV, followed by progressive worsening of his general state, culminating in death due to pulseless electrical activity.
DiscussionAmyloidosis is a systemic disease first described by Rudolph Virchow in 1854.1 It is caused by extracellular deposition of insoluble fibrils of low molecular weight proteins2,3 that form beta sheets.4 Over 30 different proteins are known to be involved in the disease,5 of which the most common forms are light-chain amyloidosis (AL), amyloid A amyloidosis (AA) and transthyretin-related amyloidosis (ATTR).6,7
Despite the heterogeneity in their structure and function, these proteins are deposited in the form of amyloid in various organs, locally or systemically, and can cause multiple organ dysfunction (Table 1).
Characteristics of different types of amyloidosis.
| Type | Precursor protein | Main organs affected | Diagnosis | Treatment | Specific characteristics |
|---|---|---|---|---|---|
| AL, primary | Light-chain amyloid | Kidney Heart CNS GIT Liver Skin | Serum and urine protein immunofixation Myelogram (5–10% plasma cells) Abdominal fat pad biopsy Bone marrow biopsy | Chemotherapy Stem cell transplantation | M/F ratio 3:2 10–15% associated with multiple myeloma |
| AA, secondary | Amyloid A | Kidney | Treat underlying disease | ||
| ATTR, hereditary (autosomal dominant) | Transthyretin, mutant | Heart Liver CNS and PNS | 99mTc-DPD scintigraphy Genetic study of transthyretin | Liver transplant Tafamidis | |
| SSA | Transthyretin, wild-type | Heart Carpel tunnel syndrome Heart | 99mTc-DPD scintigraphy Genetic study of transthyretin Linear endocardial deposits | M/F ratio 20:1 | |
| Isolated atrial amyloidosis | Atrial natriuretic peptide | Increased incidence with age and in women |
AA: amyloid A amyloidosis; AL: light-chain amyloidosis; ATTR: transthyretin-related amyloidosis; CNS: central nervous system; GIT: gastrointestinal tract; M/F: male/female; PNS: peripheral nervous system; SSA: senile systemic amyloidosis.
Cardiac amyloidosis is the result of amyloid deposits in the heart,8 the most common presentation in the West being restrictive cardiomyopathy, while in around 5% of cases, it may mimic hypertrophic cardiomyopathy.9
Cardiac involvement is found in around 50% of patients with AL amyloidosis,10 in isolation in 5% of these,11 but is rare in AA amyloidosis. ATTR amyloidosis also frequently affects the heart, although the endemic mutation in Portugal is more typically associated with neurological manifestations (familial amyloid polyneuropathy). Senile systemic amyloidosis (SSA), found predominantly in men over 70, mainly affects the heart.7
Clinical presentation varies depending on the organs involved and the degree of associated dysfunction.3 It is usually diagnosed late since symptoms are nonspecific and are often disregarded or confused with other conditions. Renal and heart failure of unknown cause are common forms of presentation, as in our patient. HF is found in around 60% of patients and is caused by deposition of amyloid fibrils between myocytes, resulting in diastolic dysfunction and hence exertional dyspnea.12 LV systolic dysfunction, peripheral edema (81%) and ascites secondary to right ventricular dysfunction are common in later stages of the disease. The most common arrhythmia is atrial fibrillation (10–15%), and chest discomfort is found in 25% of cases.10 Episodes of syncope indicate a poor prognosis, frequently culminating in sudden death.11
Unlike hypertrophic cardiomyopathy, amyloidosis is typically associated with low voltage on the ECG13 despite the presence of ventricular hypertrophy, as in the case presented. Low voltage is caused by the loss of viable myocardium due to amyloid deposits, which are electrically inert.11,14,15
Echocardiography shows diastolic dysfunction, with a restrictive pattern in 21–88% of patients.16 Speckle-tracking imaging can detect reduced LV longitudinal strain at an early stage of the disease in which ejection fraction is still preserved.11 In the case presented, there was biventricular systolic dysfunction as well as diastolic dysfunction, indicating an advanced stage of the disease (Figure 3).
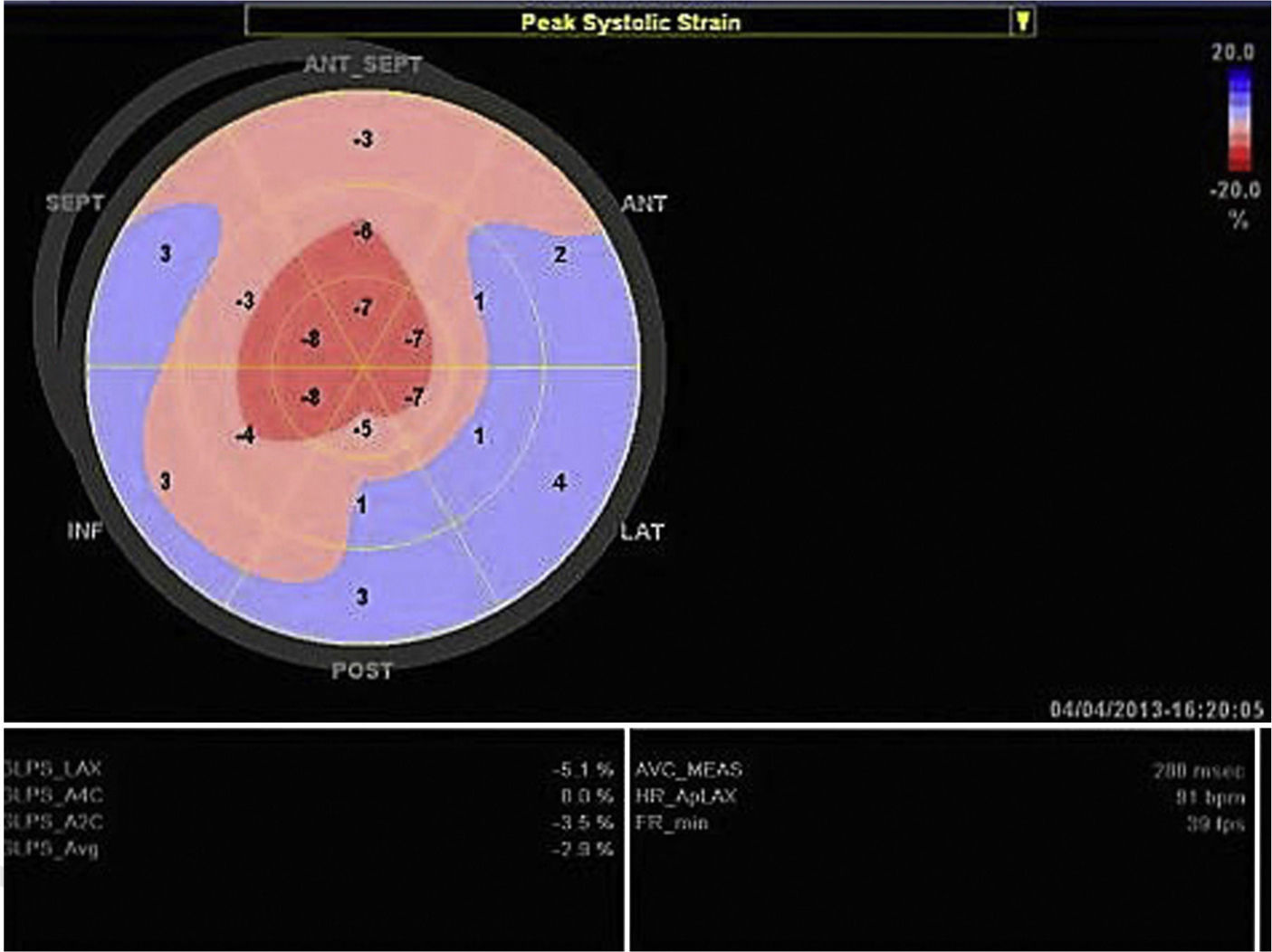
CMRI is useful for diagnosis, since it can noninvasively detect cardiac involvement in amyloidosis, with a sensitivity of 80% and specificity of 94%, by the presence of late enhancement, most commonly subendocardial and around the entire ventricular circumference.17
Biopsy of the organ involved is necessary to establish a definitive diagnosis, but abdominal fat pad biopsy is the method of choice since it is less invasive. When the sample is stained with Congo red, green birefringence is observed under polarized light (sensitivity 57–85% and specificity 92–100%). An alternative is staining with sulfated Alcian blue, which has higher specificity.14 In the case of localized cardiac amyloidosis, endomyocardial biopsy may be necessary, which is invasive, costly and not always available, but has virtually 100% sensitivity.18
The specific amyloid protein involved must be established in order to determine the appropriate treatment. In patients with cardiac amyloidosis, the most likely differential diagnosis is between AL, ATTR and SSA. Clinical differences can help in arriving at a definitive diagnosis: renal involvement is more frequent in AL, while neuropathy precedes cardiac symptoms in ATTR. Our patient had renal and heart failure as well as a history of neuropathy, and laboratory tests showed an IgG monoclonal spike, all of which pointed to AL. The myelogram and bone marrow biopsy did not reveal monoclonal plasma cells, which is the case in 10–15% of patients.7 However, it should be borne in mind that 5–10% of individuals aged over 70 have monoclonal gammopathy of undetermined significance, with a normal kappa/lambda ratio.11
The type of amyloid protein involved can be determined by immunohistochemical study of biopsied tissue (using anti-kappa, anti-lambda, anti-amyloid A and anti-transthyretin serums) or by mass spectrophotometry.
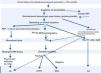
99mTc-DPD scintigraphy can detect transthyretin deposits in the heart.11 The technique is extremely useful in distinguishing between AL and ATTR, since in the latter there is selective 99mTc uptake in the heart, which is not found in AL.14 If scintigraphy detects transthyretin deposition, genetic study can be performed to distinguish ATTR (mutant transthyretin) from SSA (wild-type transthyretin) (Figure 4).
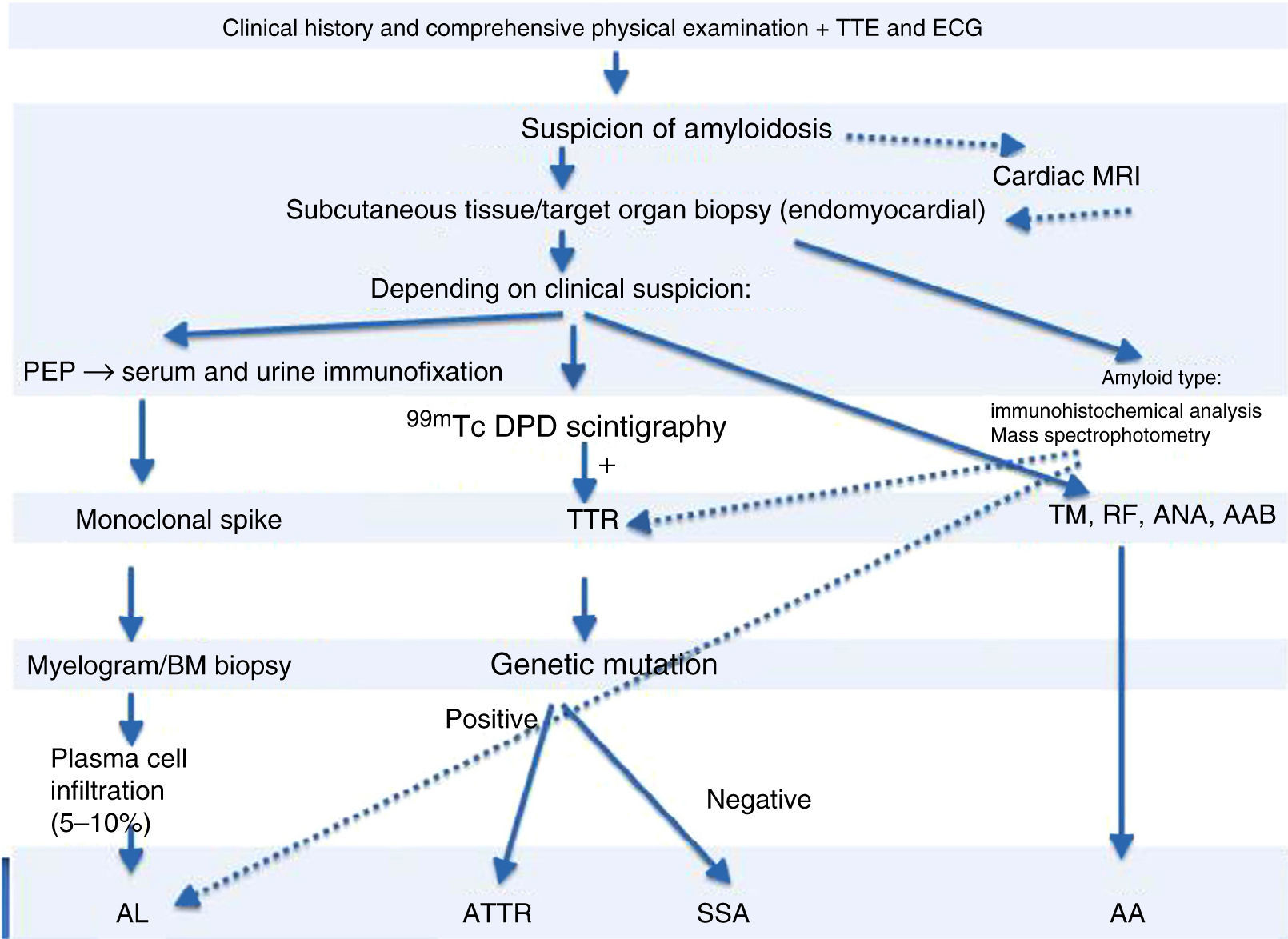
Diagnostic algorithm of cardiac amyloidosis.
AAB: autoantibodies; ANA: antinuclear antibodies; BM: bone marrow; ECG: electrocardiogram; MRI: magnetic resonance imaging; PEP: protein electrophoresis; RF: rheumatoid factor; TM: tumor markers; TTE: transthoracic echocardiogram; TTR: transthyretin.
Amyloid P scintigraphy is a noninvasive exam that provides information on the distribution and systemic extent of amyloid deposits and can also be used to assess treatment response. It cannot however detect the presence of cardiac amyloidosis due to the motion of the heart.11
Positron emission tomography, used in the evaluation of cerebral amyloidosis, is not a generally accepted method for detection of cardiac amyloidosis; however, studies have shown that it may be useful in assessing treatment response.19
Management should be coordinated by a multidisciplinary team and is mainly aimed at treatment of the underlying disease and symptom relief.
HF therapy is based on diuretics, which should be administered cautiously to prevent too steep a decrease in preload, since this can lead to a marked reduction in ventricular filling pressures and severe hypotension in patients with restrictive cardiomyopathy. Vasodilators and beta-blockers are poorly tolerated due to their hypotensive effect.20 Digoxin should be used with caution since it binds to amyloid fibrils, resulting in an increased risk of toxicity.21
The formation of atrial thrombi is common in patients with atrial fibrillation. The decision to prescribe anticoagulants should be made on an individual basis since the high risk of thromboembolism must be weighed against the increased bleeding risk due to plasma cell dyscrasia and/or renal failure.22
There is a high incidence of conduction disturbances in patients with cardiac amyloidosis, and dual-chamber pacemakers should be implanted when indicated.5,11
Heart transplantation is a controversial option due to the shortage of donors, the risk of amyloid deposition in the transplanted organ and the fact that in systemic amyloidosis the already poor prognosis is further worsened by extracardiac involvement.23 Mean survival at five years is reported as 20–30%, significantly lower than in patients transplanted for other reasons. Survival can be slightly improved (to 36%) by subsequent chemotherapy.14 Young patients with limited systemic involvement may undergo heart transplantation followed by stem cell transplantation.11
For patients with AL, the standard treatment is chemotherapy and/or autologous stem cell transplantation, depending on the disease stage and comorbidities.24
In SSA, treatment is directed at symptom relief only, while individuals under 65 may be candidates for heart transplantation.
Liver transplantation is the main treatment option for ATTR, since the altered protein is mainly produced by the liver.25 However, recurrence of extracardiac disease is often accompanied by progressive heart disease. It has been suggested that this is due to the fact that existing deposits of the mutant protein in the heart increase deposition of the normal protein through tropism. Heart and liver transplantation is recommended in patients with severe cardiac compromise. Tafamidis is available in Europe for stabilization of mutant transthyretin.
The leading causes of cardiac death are HF and sudden death due to asystole or pulseless electrical activity, against which implantable cardioverter-defibrillators are rarely effective.26
Mean survival in patients with untreated AL amyloidosis without cardiac involvement is 10–14 months; only 5% survive longer than 10 years, the extent of cardiac involvement being the main determinant of prognosis.27,28 The cause of death in around 50% of patients with AL amyloidosis is cardiac, due to arrhythmias or HF, with median survival of only six months from the onset of HF symptoms.11,27 An Italian study by Rapezzi et al. confirmed the expected differences in mortality and morbidity between AL, ATTR and SSA, patients with AL presenting a rather aggressive clinical course.28,29 The prognosis is more favorable in ATTR and SSA, irrespective of the extent of cardiac involvement.30,31
In the case of our patient, the diagnosis was made at an advanced stage of the disease. If the proteins involved have not been identified, chemotherapy is not recommended.
ConclusionsCardiac amyloidosis is rare, and diagnosis requires a high level of suspicion based on the clinical setting and the findings of noninvasive diagnostic exams, particularly transthoracic echocardiography and CMRI. A definitive diagnosis can only be made following histological study. Treatment is directed at the underlying disease and symptom relief. Prognosis is poor in AL amyloidosis with cardiac involvement.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that the procedures followed were in accordance with the regulations of the relevant clinical research ethics committee and with those of the Code of Ethics of the World Medical Association (Declaration of Helsinki).
Confidentiality of dataThe authors declare that no patient data appear in this article.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Fernandes A, Caetano F, Almeida I, Paiva L, Gomes P, Mota P, et al. Amiloidose cardíaca – abordagem diagnóstica, a propósito de um caso clínico. Rev Port Cardiol. 2016;35:305.e1–305.e7.
