





Os autores apresentam um caso de amiloidose sistémica com envolvimento cardíaco e discutem a importância de um elevado índice de suspeição para o diagnóstico, os meios de diagnóstico e as opções terapêuticas à luz do conhecimento atual. Homem de 78 anos, com antecedentes de doença renal crónica e fibrilhação auricular, admitido por insuficiência cardíaca aguda de etiologia desconhecida. O ecocardiograma transtorácico mostrou ventrículo esquerdo não dilatado, com compromisso severo da função sistólica global com overlap fenotípico de miocardiopatia hipertrófica e restritiva. Após estudo complementar alargado, o diagnóstico definitivo de amiloidose foi obtido por biópsia da gordura abdominal.
The authors present a case of systemic amyloidosis with cardiac involvement. We discuss the need for a high level of suspicion to establish a diagnosis, diagnostic techniques and treatment options. Our patient was a 78‐year‐old man with chronic renal disease and atrial fibrillation admitted with acute decompensated heart failure of unknown cause. The transthoracic echocardiogram revealed severely impaired left ventricular function with phenotypic overlap between hypertrophic and restrictive cardiomyopathy. After an extensive diagnostic workup, which included an abdominal fat pad biopsy, the final diagnosis was amyloidosis.
Homem de 78 anos, sem fatores de risco cardiovascular, com antecedentes de fibrilhação auricular paroxística e doença renal crónica (estádio IV de NKF), internado na unidade de cuidados intensivos cardíacos por insuficiência cardíaca aguda em classe de NYHA IV, de etiologia desconhecida. Da anamnese apuravam‐se queixas de dispneia de esforço e edemas periféricos com agravamento progressivo ao longo dos dois últimos meses, apresentando atualmente dispneia para esforços ligeiros, ortopneia e dispneia paroxística noturna; edemas periféricos e aumento do perímetro abdominal, perceção de diminuição do débito urinário e parestesias dos membros superiores. Ao exame objetivo apresentava‐se em anasarca, com pressão arterial 114/81mmHg, frequência cardíaca de 67bpm e saturação de O2 em ar ambiente de 90%.
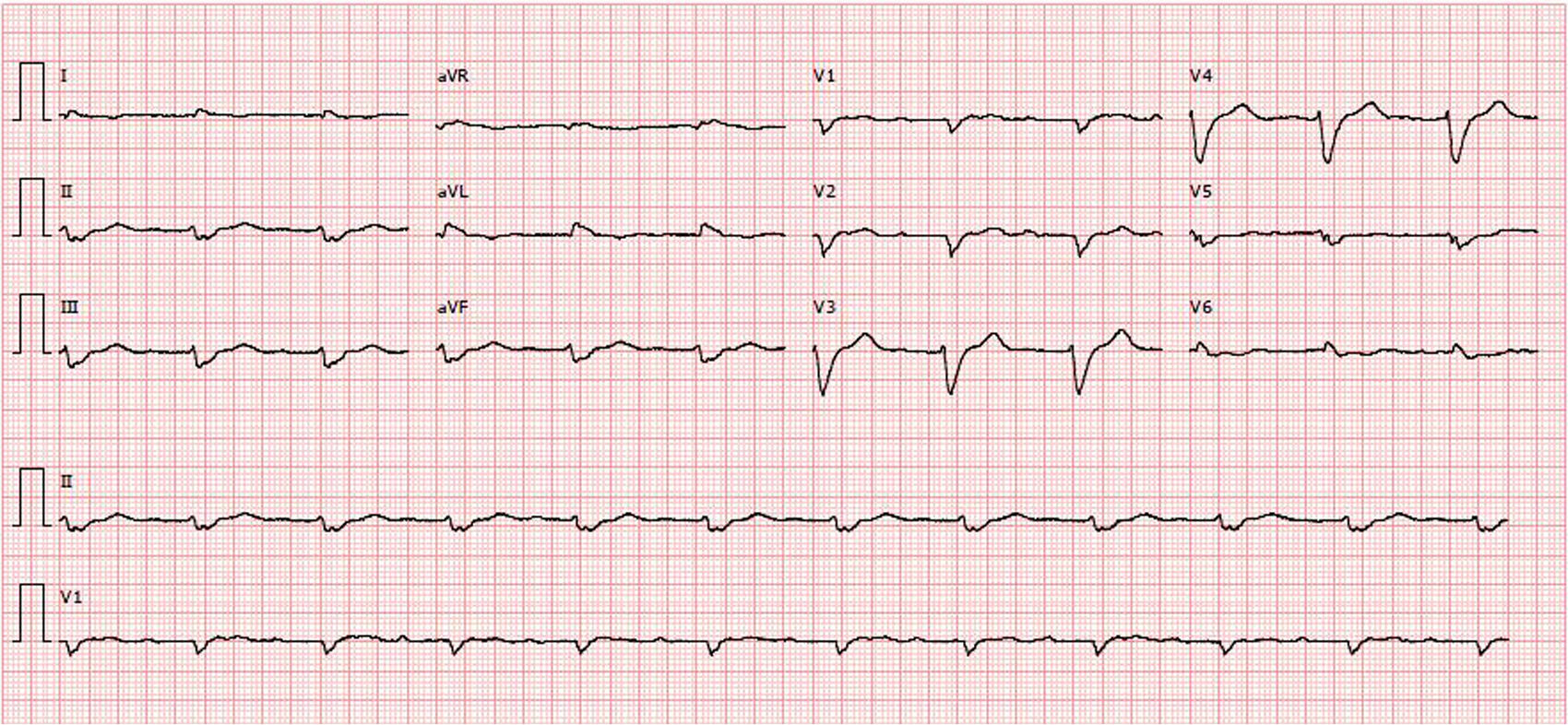
O estudo complementar realizado documentou: insuficiência respiratória tipo 2 com acidemia (pH 7,24; paO2 58mmHg, paCO2 55mmHg, HCO3 23,6mmol/l, lactatos 2,66mmol/l); anemia normocítica (Hb 12,1g/dL), sem aumento dos parâmetros inflamatórios; NT‐pro‐BNP 43300pg/ml; hiponatremia (Na+ 132,7mmol/L); doença renal crónica agudizada (ureia 24,8mmol/l e creatinina 207,8umol/l); padrão de congestão hepática (fosfatase alcalina 183U/L) com aumento das transaminases (TGO 161U/L, TGP 297U/L). A radiografia do tórax apresentava infiltrado interstício‐alveolar com padrão em «asa de borboleta». O eletrocardiograma mostrava ritmo sinusal, 67/min, bloqueio auriculoventricular do 1.° grau, baixa voltagem nas derivações do plano frontal e bloqueio completo de ramo esquerdo (Figura 1).
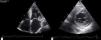
O ecocardiograma transtorácico documentou ventrículo esquerdo (VE) no limite superior do normal (diâmetro telediastólico 60mm), com paredes de ecogenicidade aumentada, hipertrofia severa do septo interventricular (20mm) e moderada das restantes paredes (parede posterior 15mm; massa do ventrículo esquerdo 200,1g/m2); dilatação biauricular severa (área da aurícula esquerda 37cm2/m2 [Figura 2 – painéis 1 e 2]); função sistólica do ventrículo esquerdo severamente comprometida (fração de ejeção de 27% pelo método de Simpson; com strain longintudinal global de ‐3%); ventrículo direito (VD) hipocontrátil (TAPSE 12mm), fluxo de enchimento ventricular esquerdo sugestivo de padrão do tipo restritivo (tempo de desaceleração de 145ms; velocidade de relaxamento diastólica precoce do anel mitral (E’) septal de 3cm/s; E’ lateral de 4cm/s; e relação E/E¿ média de 29,5); gradiente VD/AD de 43mmHg; veia cava inferior dilatada (2,3cm) e sem colapso inspiratório; derrame pericárdio circunferencial ligeiro e hipertrofia do SIA.
Não foi realizada ressonância magnética cardíaca por material protésico na anca.
Foi iniciada terapêutica anticongestiva inotrópica positiva com levosimendan e ventilação não invasiva, com progressiva melhoria clínica.
Perante os achados de hipertrofia severa do ventrículo esquerdo (VE) e eletrocardiograma com baixa voltagem, colocou‐se a hipótese de amiloidose. Foi realizada biópsia da gordura abdominal que confirmou a presença de substância amiloide, com emissão de birrefringência verde após coloração com vermelho do Congo.
Analiticamente, constatou‐se aumento da β2 microglobulina (4,98mg/L), velocidade de sedimentação de 32mm/h, proteína C reativa de 5,0mg/dl, cálcio de 2,07mmol/L, fósforo de 1,05mmol/L, paratormona de 171pg/ml, marcadores tumorais e o estudo autoimune sem alterações. A imunofixação das proteínas séricas evidenciou componente monoclonal IgG (pico de 11,8g/dl), sem alterações na imunofixação urinária. A relação entre cadeias leves kappa e lambda era de 1,22 (7,29g/dl: 5,98g/dl). Após contacto com a hematologia, foi realizado mielograma e biópsia da medula óssea que não mostraram proliferação plasmocitária. Não foram identificadas mutações nos genes que codificam as proteínas da transtirretina.
Com base nos achados foi feito o diagnóstico de amiloidose sistémica com envolvimento cardíaco. Não foi determinado o tipo de fibrilha envolvido e não foi realizada cintigrafia com tecnésio 99 difosfónico‐1,2‐propano carboxílico (Tc99m‐DPD), pelo que o diagnóstico definitivo do tipo de amiloidose não foi estabelecido.
Quatro meses depois, o doente foi novamente internado por insuficiência cardíaca descompensada em classe NYHA IV, com progressiva deterioração do estado geral que culminou em morte em dissociação eletromecânica.
DiscussãoA amiloidose é uma doença sistémica descrita pela primeira vez em 1854 por Rudolph Virchow1. É causada pela deposição extracelular de fibrilhas insolúveis de proteínas de baixo peso molecular2,3 que adquirem uma conformação de β‐pleated sheets4. Até à data, são conhecidas pelo menos 30 proteínas diferentes5, sendo as mais comuns as cadeias leves (amiloidose AL), a proteína amiloide tipo A sérica (amiloidose AA) e a transtirretina (ATTR)6,7.
Apesar da grande heterogeneidade na estrutura e função, estas proteínas depositam‐se na forma de amiloide nos vários órgãos, de forma localizada ou sistémica, podendo causar disfunção multiorgânica (Tabela 1).
Características dos diferentes tipos de amiloidose cardíaca
| Tipo | Proteína percursora | Principais órgãos envolvidos | Diagnóstico | Tratamento | Particularidades |
|---|---|---|---|---|---|
| AL, primária | Cadeias leves | Rim Coração SNC Sistema digestivo, fígado Pele | Imunofixação das Proteínas do Soro e Urina Medulograma (5‐10% plasmócito) Biópsia da gordura abdominal Biópsia da medula óssea | Quimioterapia Transplante de stem cells | 3:2homens 10‐15% associada a Mieloma Múltiplo |
| AA, secundária | Amiloide A | Rim | Tratamento da patologia de base | ||
| ATTR, hereditária (autossómica dominante) | Transtirretina mutante | Coração Fígado Sistema nervoso central e periférico | Cintigrafia Tc99m‐DPD Teste genético da transtirretina | Transplante hepático Tafamidis | |
| ASS, senil Amiloidose auricular isolada (AANF) | Transtirretina wild‐type Peptídeo natriurético auricular | Coração Síndroma túnel cárpico Coração | Cintigrafia Tc99m‐DPD Teste genético da transtirretina Depósitos lineares sob o endocárdio | Homens 20:1 Incidência aumenta com idade e no género feminino |
A amiloidose cardíaca surge da deposição de amiloide no tecido cardíaco8, sendo a forma de miocardiopatia restritiva mais frequente no mundo ocidental, podendo confundir‐se, em cerca de 5% dos casos, com miocardiopatia hipertrófica9.
O atingimento cardíaco ocorre em cerca de 50% dos casos de amiloidose AL10, dos quais 5% de forma isolada11, sendo raro na amiloidose AA. A amiloidose ATTR também afeta frequentemente o coração, apesar de a mutação endémica em Portugal cursar mais frequentemente e de forma característica com manifestações neurológicas (polineuropatia amiloidótica familiar ou PAF). A amiloidose sistémica senil é uma amiloidose essencialmente cardíaca, que surge habitualmente após os 70 anos em homens7.
A apresentação clínica é variável e relaciona‐se com o tipo de órgãos envolvidos e o grau de disfunção associado3. O diagnóstico é habitualmente tardio, uma vez que se trata de uma patologia cujas manifestações clínicas são pouco específicas, sendo frequentemente ignoradas ou confundidas com outras patologias. Tal como aconteceu no nosso caso clínico, a insuficiência renal e cardíaca de causas desconhecidas são formas de apresentação frequentes inespecíficas. A insuficiência cardíaca surge em cerca de 60% dos doentes e deve‐se ao depósito de fibrilhas amiloides entre os miócitos, que condiciona disfunção diastólica, o que se traduz em dispneia de esforço12. Em estádios mais avançados, surgem disfunção sistólica do ventrículo esquerdo e edemas periféricos (81%) e ascite secundários a disfunção do ventrículo direito. A arritmia mais frequente é a fibrilhação auricular (10‐15%). Pode surgir dor torácica em 25% dos casos10. Episódios de síncope indiciam mau prognóstico, sendo frequentemente precursores de morte súbita11.
Contrariamente à miocardiopatia hipertrófica, é característico da amiloidose ser acompanhada de baixa voltagem no eletrocardiograma13, apesar da presença de hipertrofia ventricular, tal como acontecia no nosso caso. A baixa voltagem deve‐se à perda de miocárdio viável devido aos depósitos de amiloide, proteína eletricamente inerte11,14,15.
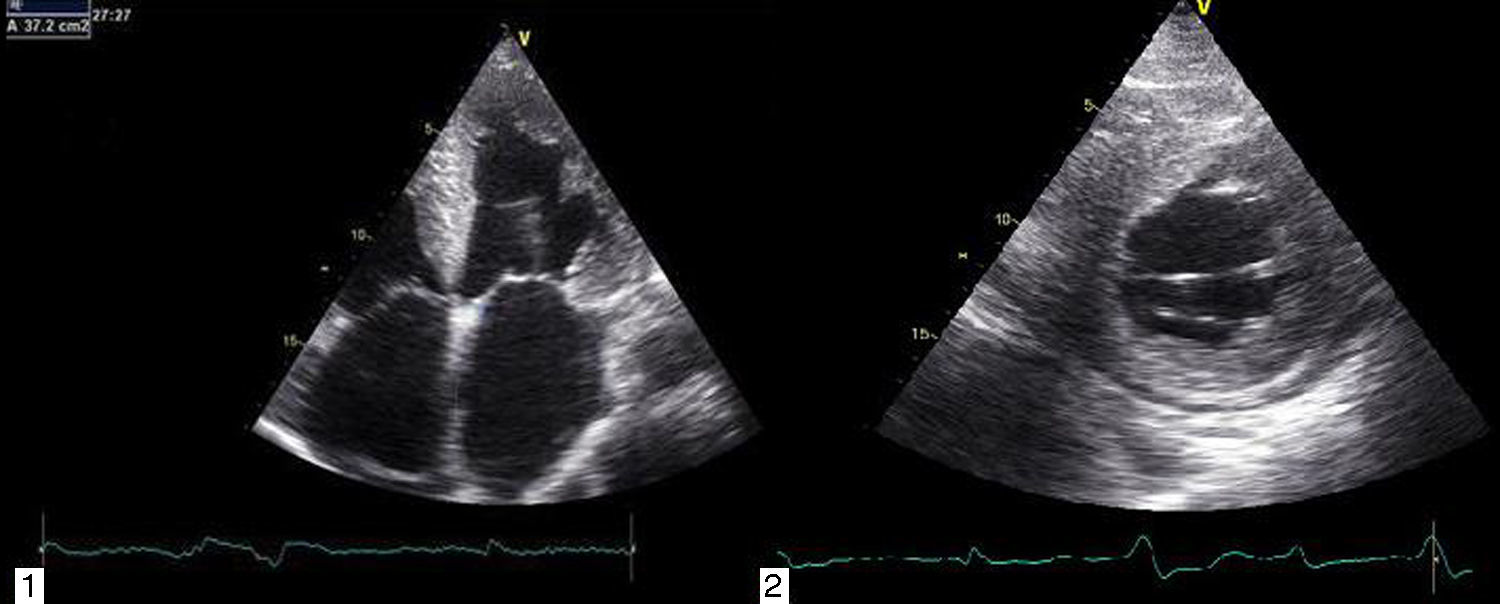
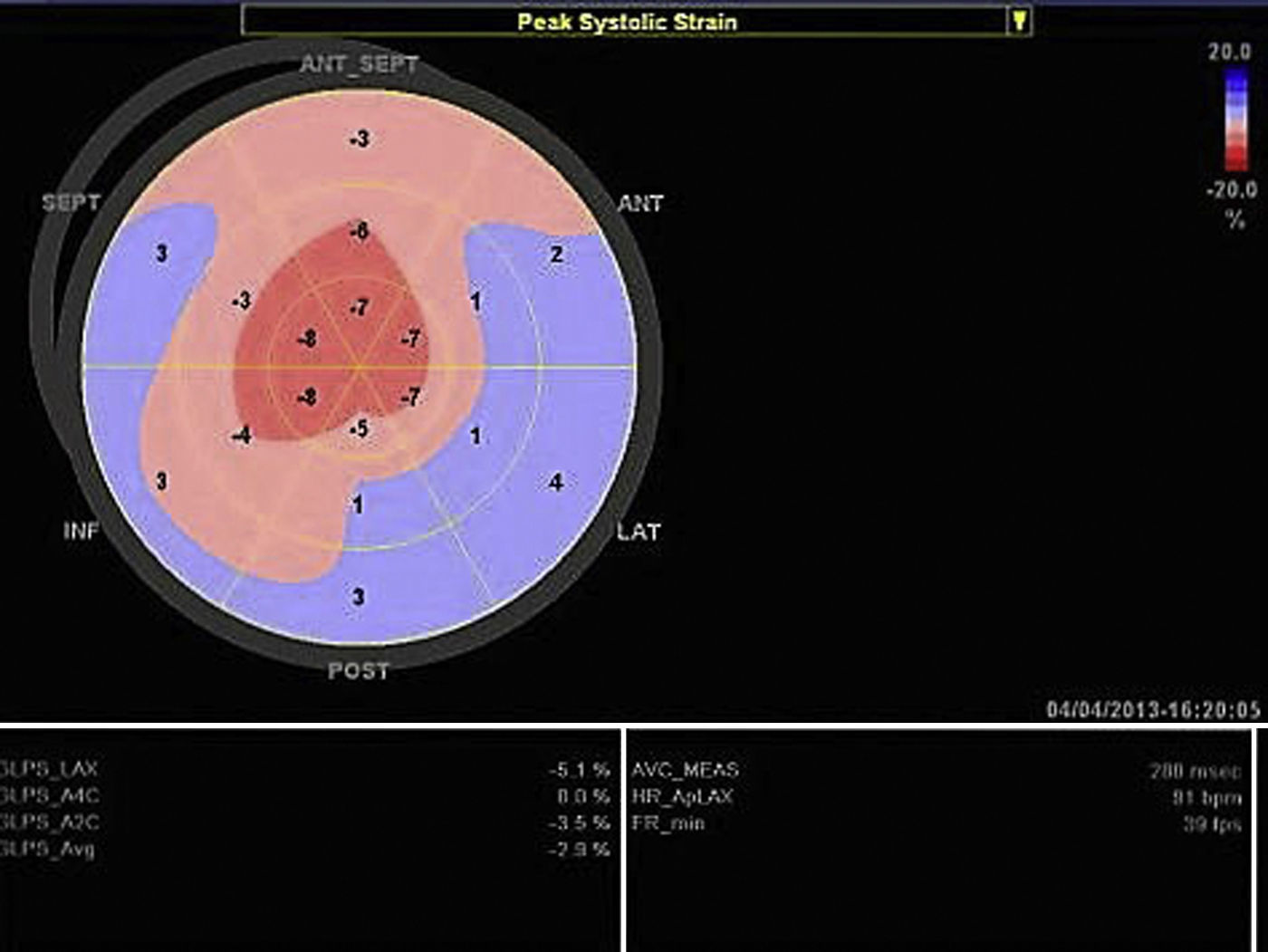
A ecocardiografia revela disfunção diastólica, com evidência de padrão restritivo em 21‐88% dos doentes16. O estudo de strain por speckle‐tracking permite detetar deformação longitudinal do VE reduzida numa fase precoce da doença, em que a fração de ejeção do ventrículo esquerdo ainda está preservada11. Neste caso existia já disfunção sistólica biventricular associada à disfunção diastólica, refletindo um estádio avançado (Figura 3).
A ressonância magnética cardíaca tem utilidade diagnóstica, uma vez que permite reconhecer de forma não invasiva, com sensibilidade de 80% e especificidade de 94%, o envolvimento cardíaco pela amiloidose traduzido pela presença de realce tardio, mais frequentemente subendocárdico e difuso em toda a circunferência ventricular17.
Para estabelecer o diagnóstico definitivo é necessária biópsia de um órgão envolvido, estando indicada para o efeito a biópsia do tecido celular subcutâneo abdominal, por ser um método menos invasivo. Ao corar a amostra com vermelho do Congo, observa‐se birrefringência verde com luz polarizada (sensibilidade 57‐85% e especificidade 92‐100%). Como alternativa, existe a coloração com sulfato azul de Alcian, técnica com maior especificidade14. No caso das amiloidoses cardíacas localizadas, poderá ser necessário realizar biópsia endomiocárdica, método invasivo, dispendioso e nem sempre disponível, com uma sensibilidade virtualmente de 100%18.
Para confirmar qual o tipo de amiloidose em causa, de forma a direcionar de forma correta o tratamento, deve ser determinado o tipo de proteína amiloide envolvida. Perante um doente com atingimento cardíaco, o diagnóstico diferencial mais provável é entre amiloidose AL, ATTR e amiloidose senil. As diferenças clínicas auxiliam‐nos a orientar o diagnóstico definitivo: o envolvimento renal ocorre mais frequentemente na amiloidose AL enquanto a presença de neuropatia precede a sintomatologia cardíaca na amiloidose ATTR. No nosso caso, o doente tinha compromisso renal e cardíaco assim como história de neuropatia; analiticamente, verificámos a presença de um pico monoclonal gamma o que, em conjunto com os dados da história, nos direcionou para a hipótese de amiloidose AL. O medulograma e a biópsia da medula óssea não mostraram uma população monoclonal de plasmócitos, o que acontece em 10‐15% dos casos7. Contudo, não podemos esquecer que em 5‐10% dos indivíduos com idade superior a 70 anos pode existir uma banda monoclonal sérica sem significado – gamopatia monoclonal de significado incerto (MGUS), sendo nestes casos a relação kappa/lambda normal11.
A determinação do tipo de proteína amiloide pode ser efetuada através de estudo imuno‐histoquímico do tecido biopsado (soros antikappa, antilambda, antiamiloide A, antitranstirretina), ou por espectrofotometria de massa.
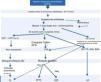
A cintigrafia com Tc99m‐DPD permite detetar a acumulação cardíaca de transtirretina11. Trata‐se de um método extremamente útil na distinção entre amiloidose AL e amiloidoses relacionadas com a transtirretina, uma vez que nestas ocorre uma captação cardíaca seletiva de Tc99m, inexistente na amiloidose AL14. Se a cintigrafia com Tc99m‐DPD detetar acumulação de transtirretina, a investigação pode ser complementada com o estudo genético da transtirretina para distinguir a amiloidose ATTR (transtirretina mutante) da amiloidose senil (transtirretina wild‐type [Figura 4]).
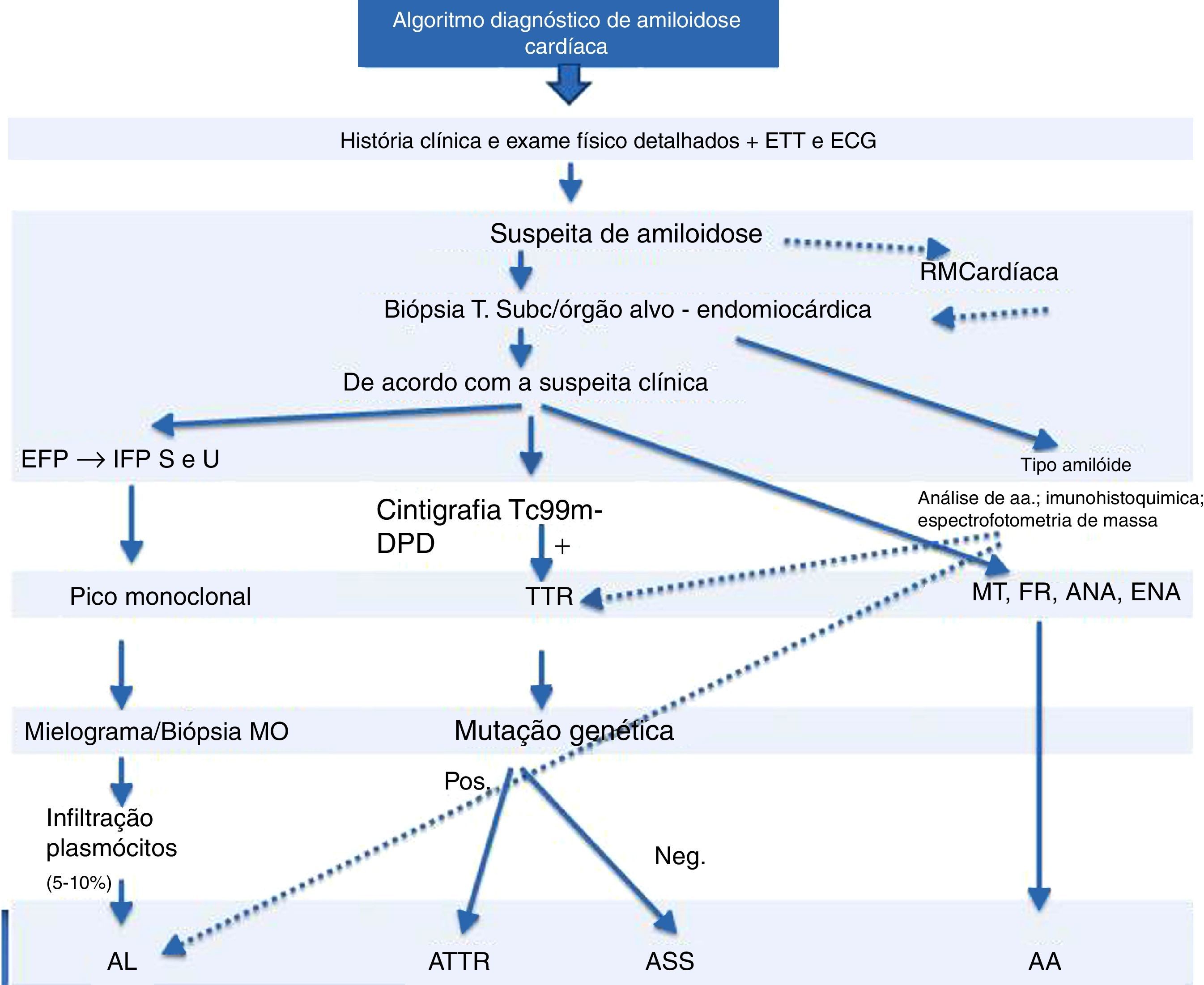
Algoritmo diagnóstico de amiloidose.
ANA: anticorpos antinucleares; ECG: eletrocardiograma; EFP: eletroforese de proteínas; ENA: autoanticorpos; ETT: ecocardiograma transtorácico; FR: fator reumatoide; IFP S e U: imunofixação soro e urina; MO: medula óssea; MT: marcadores tumorais; Neg: negativo; Pos: positivo; RM: ressonância magnética.
A cintigrafia com amiloide‐P é um exame não invasivo que dá informação em relação à distribuição, extensão sistémica dos depósitos de amiloide e seguimento da resposta ao tratamento. Não permite avaliar a presença de amiloidose cardíaca devido ao movimento do coração11.
A PET, método já utilizado para avaliar a amiloidose cerebral, não é uma forma de diagnóstico universalmente validada na deteção de amiloidose cardíaca; no entanto, existem estudos que revelam que poderá ser útil no seguimento da resposta ao tratamento19.
O tratamento tem como principais objetivos o tratamento da doença subjacente e o alívio sintomático, e deverá ser coordenado por uma equipa multidisciplinar.
O tratamento da insuficiência cardíaca assenta sobretudo em diuréticos; deve ser feito de forma judiciosa de forma a prevenir a diminuição marcada da pré‐carga, o que, em doentes com padrão restritivo, pode levar a acentuada diminuição das pressões de enchimento ventricular e hipotensão severa. Os vasodilatadores e os bloqueadores beta são mal tolerados pela hipotensão que condicionam20. O uso de digitálicos deve ser cauteloso, uma vez que as moléculas de digoxina se ligam às fibrilhas de amiloide com um risco aumentado de intoxicação digitálica21.
Num doente em fibrilhação auricular, a formação de trombos auriculares é comum. A decisão de hipocoagular deve ser tomada de forma individual, uma vez que ao elevado risco tromboembólico se contrapõe o maior risco hemorrágico, devido à discrasia sanguínea e à insuficiência renal22.
A incidência de perturbações da condução é elevada, devendo nestes doentes, quando indicado, serem implantados pacemakers de dupla câmara5,11.
A transplantação cardíaca é uma opção controversa devido à escassez de órgãos, ao risco de depósito de amiloide no órgão transplantado e ao facto de, na amiloidose com atingimento sistémico, o mau prognóstico ser condicionado pelo envolvimento extracardíaco23. Está reportada uma sobrevida média aos cinco anos de 20‐30%, significativamente inferior quando comparada com a de doentes transplantados por outros motivos. Esta sobrevida pode ser aumentada de forma discreta (36%) quando seguida por quimioterapia14. Doentes jovens com atingimento limitado poderão ser sujeitos a protocolos de transplantação sequencial cardíaco e de stem‐cells11.
Nos doentes com amiloidose AL, o tratamento standard, dependendo do estádio e das comorbilidades existentes, é a quimioterapia e/ou transplante autólogo de stem‐cells24.
Na amiloidose senil, o tratamento é apenas sintomático, podendo indivíduos mais jovens (<65 anos) ser candidatos a transplantação cardíaca.
A transplantação hepática é a base do tratamento da amiloidose ATTR, uma vez que a proteína alterada tem síntese maioritariamente hepática25. No entanto, a regressão da doença extracardíaca é frequentemente acompanhada pela progressão da cardiopatia. Postula‐se que isto se deve ao facto da proteína mutante já depositada a nível cardíaco aumentar o tropismo para a deposição de proteína normal. Nos doentes com compromisso cardíaco severo é advogada a opção transplante coração‐fígado. Existe já disponível na Europa uma molécula estabilizadora da transtirretina nativa: o tafamidis.
As principais causas de morte cardíaca são a insuficiência cardíaca e a morte súbita por assistolia ou atividade elétrica sem pulso, sendo os cardiodesfibrilhadores implantáveis raramente eficazes26.
Na amiloidose AL, a sobrevida média nos doentes sem envolvimento cardíaco, não tratados, é de 10‐14 meses; apenas 5% sobrevive aos dez anos, sendo a extensão do atingimento cardíaco o principal determinante do prognóstico27,28. Em cerca de 50% dos doentes com amiloidose AL a causa da morte é cardíaca, por disritmias ou insuficiência cardíaca, sendo a sobrevida média de cerca de seis meses após o início da clínica da insuficiência cardíaca11,27. Um estudo italiano de Rapezzi et al. comprovou as esperadas diferenças em relação à mortalidade e morbilidade entre doentes com amiloidose AL, ATTR e senil, verificando‐se um curso muito mais agressivo nos doentes com amiloidose AL28,29. Na amiloidose ATTR como na ASS, o prognóstico é muito mais favorável, independentemente do grau de envolvimento cardíaco30,31.
No caso particular do nosso doente, o diagnóstico foi realizado numa fase muito avançada da doença. Na ausência de identificação das proteínas envolvidas também não se recomenda a quimioterapia.
ConclusãoA amiloidose cardíaca é uma patologia rara. O diagnóstico exige um elevado índice de suspeição com base na clínica e nos achados dos exames complementares não invasivos, particularmente o ecocardiograma transtorácico e a ressonância cardíaca. O diagnóstico final exige sempre demonstração histológica. O tratamento é dirigido à doença subjacente e ao alívio dos sintomas. O prognóstico é sombrio nos casos de amiloidose AL com envolvimento cardíaco.
Responsabilidades éticasProteção de pessoas e animaisOs autores declaram que para esta investigação não se realizaram experiências em seres humanos e/ou animais.
Confidencialidade dos dadosOs autores declaram ter seguido os protocolos do seu centro de trabalho acerca da publicação dos dados de pacientes.
Direito à privacidade e consentimento escritoOs autores declaram ter recebido consentimento escrito dos pacientes e/ou sujeitos mencionados no artigo. O autor para correspondência deve estar na posse deste documento.
Conflito de interessesOs autores declaram não haver conflito de interesses.
Agradece‐se à Dr.a Olga Azevedo pela ajuda prestada.
