







Genetics has assumed a pivotal role in clarifying the pathophysiology of cardiomyopathies, facilitating molecular diagnosis, and enabling effective family screening. The advent of next-generation sequencing has revolutionized genetic testing by enabling cost-effective, high-throughput analysis. It is imperative for cardiovascular physicians to mainstream genetic testing into their clinical decision-making. Although a definitive genotype-phenotype correlation may not always be evident, several genotypes have emerged as valuable risk predictors for disease severity and progression. European guidelines emphasize the importance of genetic tests for predicting clinical outcome in cardiomyopathies. While further research is essential to bridge existing gaps in the genetic evidence on cardiomyopathies, there is considerable potential for significant advancements.
A genética assumiu um papel fundamental na clarificação da fisiopatologia das miocardiopatias, facilitando o diagnóstico molecular e possibilitando o rastreio familiar eficaz. O desenvolvimento da sequenciação de nova geração revolucionou os testes genéticos, permitindo análises de alto rendimento a custos acessíveis. Atualmente, a incorporação dos testes genéticos no processo de tomada de decisões clínicas é essencial. Embora nem sempre exista uma correlação genótipo-fenótipo reproduzível, diversos genótipos surgiram como preditores de risco para a gravidade e progressão da doença. As guidelines europeias advogam o uso de testes genético nas suas recomendações, demonstrando notáveis vantagens na previsão de outcomes clínicos. Embora sejam necessárias mais estudos para preencher as lacunas na compreensão da genética associada às miocardiopatias, o potencial para avanços é vasto.

Cardiomyopathies constitute a heterogeneous group of diseases, in which pathophysiology, phenotypes, management, and life-threatening arrhythmic risk vary widely.1 They are characterized by structural and functional abnormalities in the heart muscle that is not explained by coronary or valvular disease or abnormal loading conditions. Overall, they carry an increased and variable risk of heart failure progression and arrhythmic events. While environmental factors and lifestyle choices play a role, it is becoming increasingly evident that genetics is crucial to understanding the underlying mechanisms of cardiomyopathies.2
In 2017, a new system for classifying cardiomyopathies, including genetic background, was proposed, under the acronym MOGES. This system brought together the genetic origins of the disease and its clinical and pathophysiological characteristics, but its adoption has been limited due to its complexity, which renders it impractical for widespread use.3 Overall, the need remains for alternative methods of including genetics in the approach to cardiomyopathies.
While a consistent correlation between genotype and phenotype cannot always be established, particular genes are known to be associated with rapid progression to heart failure, greater arrhythmic risk, or a more inflammatory profile.4
Hypertrophic cardiomyopathy (HCM) is arguably one of the most compelling examples of the influence of genetics, especially in the field of differential diagnosis. The identification of phenocopies through genetic testing enables targeted therapeutic management, impacting prognosis.5
In dilated cardiomyopathy (DCM), genetic testing has enabled identification of an underlying causative monogenic variant in 30–35% of individuals. Classification based on genotype was discovered to be especially effective in predicting the likelihood of sudden cardiac death (SCD) and identified the variants associated with worse outcomes, such as in the LMNA gene.6 Moreover, genetic characterization is a pillar in the diagnosis of neuromuscular and mitochondrial disease.7,8
In this review, we aim to offer clinicians a targeted, evidence-based guide to performing genetic testing as a valuable tool to improve the management of inherited cardiomyopathies. The review is divided into two parts: the approach to HCM, non-dilated left ventricular cardiomyopathy and DCM (Part 1), and the approach to syndromic cardiomyopathy and phenocopies (Part 2). This manuscript constitutes Part 1.
Genetic testing for inherited cardiomyopathiesFamilial forms of cardiomyopathies are associated with different modes of inheritance. The major focus for inherited cardiomyopathies is on Mendelian disease genes, for which the inheritance patterns may be autosomal dominant, autosomal recessive, X-linked, or mitochondrial.9
The ultimate goal of genetic testing in inherited cardiomyopathies is to improve patient outcomes. The identification of genetic variations is a critical step toward precision medicine and advancing patient care. Genetic information helps clinicians to make an accurate diagnosis, tailor treatments, and ultimately improve patient prognosis.10
Sanger sequencing, which reads the sequence of a small, targeted region of the human genome, has its limitations, particularly low throughput and high costs. While no longer the primary tool for genetic diagnosis, it is still a powerful and cost-effective method, especially for targeted variant analysis. It is particularly useful for family cascade screening, identifying causative genetic variants previously identified in relatives of index patients.11
The limitations of Sanger sequencing ultimately led to new and more advanced sequencing technologies. Next-generation sequencing (NGS) revolutionized DNA sequencing by enabling the parallel processing of thousands to millions of DNA fragments simultaneously, significantly increasing the speed and scale of genetic analysis. This high-throughput sequencing method, which offers high customizability, enabling tailoring to specific testing objectives, has become the cornerstone in numerous diagnostic contexts. NGS can be applied in various settings, such as targeted sequencing for focused multigene panel analysis including mitochondrial DNA (mtDNA) analysis, whole-exome sequencing for protein-coding exons, and whole-genome sequencing for comprehensive DNA sequences, encompassing both coding and non-coding regions.12
The continuous improvement of computational tools and NGS methods enables the detection of copy-number variants (CNVs), including small single-exon deletions or duplications, as well as the detection of single-nucleotide variants (SNVs). However, alternative non-sequencing quantification approaches such as multiplex ligation-dependent probe amplification remain the most reliable methodology to identify deletions and duplications.13
When choosing the genetic analysis technique, consideration should be given to the balance between its cost and its thoroughness, including horizontal and vertical coverage of the regions of interest, the sensitivity and specificity of the CNV analysis, the possibility of mtDNA analysis, or even subsequent reanalysis of data according to evolution of patient phenotype or changes in family history.13
The adoption of NGS techniques in diagnostic laboratories, which enable a broader analysis of genes, has led to improved identification of variants in genes linked to various disorders, thereby introducing an additional layer of complexity to variant analysis. This abundance of data has created the need for a standardized method for interpreting and classifying variants. To meet this need, in 2015 the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) published general guidelines for the classification of genetic variants.14 These guidelines cover both laboratory-based evidence and clinical data, including functional assays, segregation analysis, population frequency data, and computational predictions, to establish a tiered system that assesses specific criteria to determine the likelihood of a variant's pathogenicity. Twenty-eight criteria were defined by the ACMG/AMP guidelines to standardize the classification of each variant. Based on the available evidence, variants can be divided into five categories: pathogenic (P), considered to have a high likelihood of contributing to disease; likely pathogenic (LP), for which there is substantial evidence suggesting association with disease, though not sufficiently robust to be considered a definite pathogenic variant; uncertain significance (US), lacking evidence to be confidently classified as either disease-causing or benign; likely benign (LB), for which evidence exists indicating it is highly unlikely to contribute to disease; and benign (B), for which there is strong evidence indicating that it is not associated with disease or phenotype. Only pathogenic or likely pathogenic variants can be used in clinical settings, for personalized management or predictive testing. Accurate classification is crucial to providing appropriate genetic counseling and determining optimum medical management.14
While the 2015 ACMG/AMP guidelines serve as a valuable foundation for the classification of genetic variants, providing a standardized framework for variant classification, they were designed to address all Mendelian disorders. The complexity and context-specific nature of genetic diseases often require tailored guidelines to ensure well-defined and clinically important results, cardiomyopathies being no exception, so there is a growing recognition that disease- and gene-specific guidelines are essential for more accurate and nuanced interpretation of variants.14
To address this issue, in 2018 ClinGen's Inherited Cardiomyopathy Expert Panel published specific classification guidelines for MYH7 variants.15 For the seven other sarcomeric genes, specific guidelines are currently under review (ACTC1, MYL2, MYL3, TNNI3, TNNT2 and TPM1) or in preparation (MYBPC3).16
Hypertrophic cardiomyopathyHCM is the most common inherited heart disease, with an estimated prevalence between 1:250 and 1:500 individuals, and is defined by unexplained isolated myocardial hypertrophy, with frequent concomitant dynamic left ventricular outflow tract (LVOT) obstruction, diastolic ventricular dysfunction and reduced ejection fraction.17 Its histopathology is characterized by myocyte disarray, coronary microvascular dysfunction, and fibrosis.18
HCM primarily follows an autosomal dominant inheritance pattern. The yield of genetic testing in HCM using NGS is 30–60%, and the likelihood of obtaining a positive genetic test is greater when there is a positive family history of HCM.19 Its phenotypic expression displays notable diversity in the extent of hypertrophy and clinical course, even within families carrying identical variants; it is not uncommon to find patients sharing the same genotype yet manifesting different disease phenotypes. This variability ranges from asymptomatic individuals to those with severe arrhythmias and progression to heart failure, and arises at least partially from the interplay between the causal variant and other genetic, epigenetic, and environmental factors. Although autosomal recessive and X-linked modes of inheritance have been described, they are uncommon. X-linked inheritance should raise suspicion of a phenocopy condition.20
HCM patients can be classified into three distinct populations based on genetic etiology. The first is the sarcomere variant-positive (SARC+) group, encompassing individuals carrying variants in sarcomeric genes. The second is the sarcomere variant-negative (SARC−) population, consisting of patients with no identifiable causal variants, while the third group consists of phenocopies – conditions sharing the clinical phenotype of HCM but characterized by different pathophysiology, and notably requiring specific therapeutic strategies.20
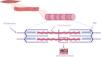
Sarcomeric hypertrophic cardiomyopathySarcomeric HCM is the main cause of familial HCM and results from P/LP variants in genes encoding protein constituents of sarcomeres. These structures constitute the fundamental contractile units of myocytes, generating the force behind muscle contraction. This category includes a diverse array of genes (Table 1 and Figure 1).21
Hypertrophic cardiomyopathy: disease-causing genes.
| Category | Gene (frequency) |
|---|---|
| Thick filament sarcomere proteins | MYH7 (33%), MYBPC3 (50%), MYL2 (<3%), MYL3 (<3%) |
| Thin filament sarcomere proteins | TNNT2 (4%), TNNI3 (5%), TNNC1, TPM1 (<3%), ACTC1 (<3%) |
| Proteins regulating calcium homeostasis in cardiac muscle | PLN (<3%) |
| Sarcomere-associated proteins | ALPK3 (rare) |

Within the cluster of genes encoding thick filament protein components, MYH7 and MYBPC3 are the most common causal genes. These genes encode beta-myosin heavy chain and myosin-binding protein C, respectively, and together are responsible for around 40% of all cases of HCM. Pathogenic variants in MYH7 are primarily missense variants. In the MYBPC3 gene, there is an over-representation of truncating variants, such as indels and splicing and nonsense variants. MYL2 and MYL3 are responsible for fewer than 1–3% of HCM cases, making them rare contributors.
Thin filament sarcomere proteinsGenes encoding thin filament protein components are responsible for around 5% of HCM cases. Many pathogenic variants in the TNNT2 gene, mainly missense, have been identified in patients with HCM.22
Other sarcomere componentsOccasionally, variants in genes coding for proteins with structural or enzymatic roles located within other components of the sarcomere have been identified as genetic triggers for SARC+ HCM. However, these underlying causes are less common.20
Determinants of hypertrophic cardiomyopathy phenotypeWithin genetic determinants, different factors can contribute to shaping the phenotype. Modifier genes, which affect the causal variant, as well as polymorphic variations in different genes, have demonstrated an effect on wall thickness. Furthermore, the presence of multiple genetic variants, such as double heterozygous, compound heterozygous, and homozygous variants, may correlate with increased disease severity.23
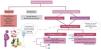
Different aspects of sarcomere-positive vs. genotype-negative hypertrophic cardiomyopathyPatients diagnosed with SARC+ HCM typically exhibit an earlier onset and a greater frequency of positive family history than those with SARC− HCM (Figure 2). On the other hand, individuals with SARC− HCM present an increased prevalence of cardiovascular risk factors, including obesity and hypertension, implying that the development of hypertrophy may also be influenced by age and changes associated with cardiovascular risk factors such as pressure overload from hypertension, obesity, and the metabolic impact of diabetes.20

SARC+ patients have been shown to have an increased risk of death and progression to heart failure. These patients present with greater wall thickness, higher rates of non-sustained ventricular tachycardia (NSVT), and consequently higher SCD risk scores as assessed by the European Society of Cardiology (ESC)’s HCM Risk SCD score, a greater late gadolinium enhancement (LGE) burden and increased extracellular volume. SARC− patients, who are predominantly male, are at the other end of this spectrum, being less likely to have indication for an implantable cardioverter defibrillator (ICD).24
The presence of reverse septal curvature is more common in SARC+ and is a strong predictor of a positive genetic test. Notably, this may explain the higher proportion of patients with a significant LVOT gradient in the SARC− subgroup compared to the SARC+ group.20
Varying impacts of different sarcomere variantsVariants in genes encoding thin myofilament proteins, such as TNNT2, have different effects on the phenotype from those affecting thick myofilaments. Alterations in thin filament proteins result in less severe cardiac hypertrophy but carry an elevated risk of systolic dysfunction. However, susceptibility to serious cardiac arrhythmias and SCD appears to be unaffected, regardless of whether the variants affect thin or thick filaments.25 Notably, genetic variations also correlate with atrial fibrillation (AF). Among patients with sarcomeric variants, those in the MYH7 gene are associated with a higher incidence of AF, even after accounting for clinical and echocardiographic factors.26
PhenocopiesPhenocopies are conditions that mimic the phenotypic traits of HCM but diverge significantly in terms of their underlying pathophysiology and therapeutic approaches. Recognition of phenocopies, which constitute 3–5% of cases involving unexplained left ventricular hypertrophy (LVH), stands to profoundly influence patient prognosis by facilitating targeted interventions.21 Within this category are conditions such as lysosomal and glycogen storage disorders, transthyretin cardiac amyloidosis and RASopathies, which are covered in the second part of this review.
Dilated cardiomyopathyDCM is characterized by left ventricular dilatation and systolic dysfunction, resulting from remodeling and fibrosis. Defects in cytoskeletal proteins can predispose to DCM by reducing force transmission and/or resistance to mechanical stress.27 Pathophysiological changes ultimately lead to a decrease in stroke volume and cardiac output, with impaired ventricular filling and increased end-diastolic pressure.
Although heart failure therapies have made impressive progress in recent decades, DCM mortality rates remain high, and it is one of the leading reasons for heart transplantation.28 Mortality in DCM is mostly due to advanced heart failure or SCD.29 DCM has a wide variety of causes, often classified as genetic or acquired disorders. A positive family history can be identified in 30–50% of DCM cases, and a causative genetic variant can be identified in up to 40% (Table 2).30
Genes associated with non-syndromic dilated cardiomyopathy.
| Gene | Inheritance | Frequency | Features | Other phenotypes |
|---|---|---|---|---|
| TTN | Dominant | 15%–20% | DCM is associated with truncating variants | LGMD2J; hereditary myopathy with early respiratory failure; Udd distal myopathy–tibial muscular dystrophy; Salih myopathy; HCM |
| LMNA | Dominant | 6% | Prominent conduction disease; almost complete penetrance by the age of 70 | Emery-Dreifuss muscular dystrophy; Hutchinson-Gilford progeria syndrome; NDLV |
| MYH7 | Dominant | 4% | DCM | Laing distal myopathy; HCM; myosin storage myopathy; scapuloperoneal myopathy |
| FLNC | Dominant | 2%–4% | Arrhythmia; CMR fibrosis with subepicardial ring-like pattern | Myofibrillar myopathy; HCM; RCM; distal myopathy; LVNDC |
| BAG3 | Dominant | 3% | DCM | Myofibrillar myopathy |
| TNNT2 | Dominant | 3% | DCMAssociated with ventricular arrhythmias | HCM; RCM |
| RBM205 | Dominant | 2% | Rapid progression: SCD and end-stage heart failure are common, high incidence of ventricular arrhythmias | NDLVC |
| SCN5A | Dominant | 2% | Arrhythmia/conduction system disease; early onset | Long QT syndrome; Brugada syndrome; idiopathic ventricular fibrillation, NDLVC |
CMR: cardiac magnetic resonance; DCM: dilated cardiomyopathy; HCM: hypertrophic cardiomyopathy; LGMD2J: limb-girdle muscular dystrophy type 2J; LGMDR: autosomal recessive limb-girdle muscular dystrophy; MT: Charcot-Marie-Tooth hereditary neuropathy; NDLVC: non-dilated left ventricular cardiomyopathy; RCM: restrictive cardiomyopathy.
Shaded light pink cells represent genes also associated with non-dilated left ventricular cardiomyopathy.
Variants in TTN are the most common genetic cause of DCM, and are associated with familial DCM in around 20% of cases. Titin, the protein encoded by the TTN gene, is the largest protein in the human body. It connects the Z disc to the M line in the sarcomere (Figure 1). Many of the DCM-causing TTN variants are heterozygous truncating variants (TTNtvs) that include frameshift, nonsense, and essential splice site variants and are over-represented in the A-band titin segment. Not all individuals who carry a TTNtv develop DCM, and a multifactorial disease model has been proposed for the development of a TTNtv-based phenotype.32,33 The phenotypic expression of DCM starts from around 40 years of age and is sex-dependent.34,35 Phenotype seems to depend not only on the penetrance or malignancy of the mutated gene, but also on epigenetics, age, toxic factors, pregnancy, and a variety of acquired diseases.36 Genetic determinants often increase susceptibility or act as modifying factors in the presence of an external cause for DCM.
The clinical manifestations associated with TTNtvs include severe left ventricular dilatation and systolic dysfunction. These manifestations often show a positive response to optimal medical treatment, resulting in a high frequency of left ventricular reverse remodeling and outcomes that closely resemble those observed in individuals with DCM. Nevertheless, over the long term, there is a risk of progressive deterioration in left ventricular systolic function and end-stage heart failure.37–39 Regarding arrhythmic risk, recent evidence suggests that TTNtv DCM is associated with frequent atrial and ventricular arrhythmias, with almost one-third of patients developing atrial arrhythmias and one-half NSVT. Malignant arrhythmic events tend to be more frequent in patients with severe left ventricular systolic dysfunction, which explains the current ejection fraction threshold of less than 35% for the placement of an ICD as primary prevention. Overall arrhythmic risk in TTNtv DCM seems to be greater than in idiopathic DMC, while arrhythmic outcomes are milder than with other more arrhythmogenic genes like LMNA, FLNC, PLN, and RBM20.40–42
The ECG is an essential tool in the diagnosis and risk stratification of DCM. Associations of pathogenic variants with ECG findings have been investigated by multiple studies. Patients with LMNA variants tend to manifest early conduction abnormalities, including sinus dysfunction, atrioventricular conduction block, bundle branch block or hemiblock, significantly more often than those with PLN, RBM20 or MYBPC3 variants. Regarding TTN variants, studies have failed to show a typical or common ECG pattern. However, recent evidence suggests that patients with TTNtvs have a higher prevalence of AF, while the prevalence of left bundle branch block and conduction abnormalities is lower than in other variants.40,43,44
While genetic variants are typically more prevalent in familial DCM cases, they can still be identified in over 20% of non-familial cases. Identification of a pathogenic variant in non-familial DCM brings significant benefits, as it can influence decisions regarding device implantation, offer valuable insights for genetic counseling, and facilitate the screening of family members for potential risks. It is important to note that the patient's age should not be an obstacle to genetic testing in this context. In resource-constrained settings, considering the adoption of specialized scoring systems designed to identify DCM patients with a high likelihood of possessing a positive genotype, such as the Madrid DCM Genotype Score (https://madridDCMscore.com), may be a strategic approach to prioritize genetic testing. Furthermore, it is worth mentioning that factors such as alcohol consumption, chemotherapy, and pregnancy can act as triggers for DCM in patients with an underlying pathogenic variant. Therefore, these patients may also derive benefits from genetic testing (Figure 3).1

While genetic DCM primarily affects the heart in most cases, it is noteworthy that several syndromic genetic conditions include DCM as one of their features, for which genetic testing plays an important diagnostic role. This group of diseases is addressed in a separate section.
Non-dilated left ventricular cardiomyopathyNon-dilated left ventricular cardiomyopathy (NDLVC) is characterized by the presence of non-ischemic scarring or fatty tissue replacement in the left ventricle, without left ventricular dilatation. This newly defined condition includes individuals who were formerly diagnosed with arrhythmogenic left ventricular cardiomyopathy and arrhythmogenic cardiomyopathy (ACM). The most frequently implicated genes in NDLVC are DSP (desmoplakin), FLNC (involving truncating variants), DES (desmin), LMNA, and PLN (phospholamban).1 However, it is important to note that there is considerable genetic overlap with both DCM and arrhythmogenic right ventricular cardiomyopathy (ARVC), as outlined in Tables 2 and 3.
Genes associated with arrhythmogenic right ventricular cardiomyopathy and their clinical relevance.
| Gene | Inheritance | Frequency | Clinical/imaging remarks |
|---|---|---|---|
| PKP2 | Dominant | 34%–74% | Particularly prominent in athletes |
| DSG2 | Dominant | 5%–26% | |
| DSP | Dominant | 2%–15% | Frequently presents as myocarditisAssociated with hot phases; associated with NDLVCSubepicardial, ring-like LGE pattern |
| DSC2 | Dominant | 1%–2% | ARVC can be non-syndromic or associated with mild palmoplantar keratoderma and woolly hair |
| JUP | Both | 0.5%–2% | NAXOS syndrome, but also in non-syndromic individuals |
| DES | Dominant | Rare | Conduction system disease and dilated cardiomyopathy. Associated with Kaiser-type neurogenic scapuloperoneal syndrome |
| TMEM43 | Dominant | Rare | Auditory neuropathy and myopathy commonly present, associated with NDLVCSubepicardial, ring-like pattern fibrosis in CMR |
ARVC: arrhythmogenic right ventricular cardiomyopathy; HF: heart failure; LGE: late gadolinium enhancement; NDLVC: non-dilated left ventricular cardiomyopathy; SCD: sudden cardiac death. Shaded light pink cells indicate genes also associated with non-dilated left ventricular cardiomyopathy.
Correlation with ECG changes can raise suspicion of specific pathogenic variants. A substantial proportion of individuals carrying pathogenic LMNA variants (laminopathies) exhibit a prolonged PR interval, along with various arrhythmias including supraventricular tachycardia, atrial fibrillation, atrial flutter, and premature ventricular contractions. By contrast, patients with FLNC-related conditions often present with mild conduction abnormalities, significantly reduced QRS voltage in the limb leads and T-wave inversion, particularly in the lateral leads. Truncating FLNC variants are associated with a phenotype that includes dilated cardiomyopathy and a high risk of premature sudden death.45
Specific risk prediction scores have been formulated to include genotype in the decision-making process for ICD placement. Examples of such scores include the LMNA risk score (https://lmna-risk-vta.fr)541 and the PLN p.Arg14del variant risk calculator (https://plnriskcalculator.shinyapps.io/final_shiny). Whenever these scores are accessible, they should be employed to inform the decision-making process for primary prevention ICD placement, in conjunction with shared decision-making discussions.1
Arrhythmogenic right ventricular cardiomyopathyARVC is an inherited heart muscle disease that is characterized by progressive replacement of the ventricular myocardium with fibrofatty tissue.46 Fibrofatty replacement typically occurs from the subepicardium toward the subendocardium,47 predisposing patients to progressive ventricular dysfunction, malignant ventricular arrhythmias and SCD.
The pathophysiology of ARVC is mainly related to genetically abnormal desmosomal proteins, leading to myocyte detachment and death. The desmosome complex consists of desmoplakin (DSP) (the intermediate filament holder), transmembrane proteins such as cadherins (desmocollin-2 [DSC2] and desmoglein-2 [DSG2]), and linkage proteins (including plakophilin-2 [PKP-2] and plakoglobin [JUP], which connect DSP and cadherin tails. Desmosomes are vital in mechanically stressed tissues such as those in the heart, enabling cell-to-cell adhesion and force transmission between the junctional complex and the cytoskeleton. Variants have also been found in non-desmosomal proteins linked to the cytoskeleton, sarcoplasmic reticulum, sarcomere, ion channels and cytoskeleton (including transmembrane protein 43 (TMEM43), DES, FLNC, and overlappingly DSP). The main genes related to ARVC and their clinical relevance are summarized in Table 3.48,49
These structural changes result in various clinical phenotypes, with desmosomal protein variants often associated with right ventricular involvement and non-desmosomal variants linked to left or biventricular involvement, though overlap is common. Inheritance is typically autosomal dominant with incomplete penetrance and variable expressivity, which is strongly influenced by exercise intensity.52
Moreover, inflammation plays a significant role in the pathophysiology and clinical presentation of this condition. Disease progression is not a continuous process; instead, it occurs intermittently in episodes characterized by increased inflammatory activity, referred to as hot phases. These episodes may be accompanied by an increase in arrhythmic episodes. Notably, variants in DSP have been associated with susceptibility to these inflammatory processes.53
DiscussionWe have highlighted the central contributions of genetics to the different types of cardiomyopathy, demonstrating its pivotal role across the entire spectrum of patient care. Genetics not only complements but also shapes the diagnosis, management, and prognosis in cardiomyopathy cases. Consequently, a shift toward embracing a genotype-based classification system for cardiomyopathies is becoming increasingly evident.3 The latest ESC guidelines for the management of cardiomyopathies (2023) emphasize the significance of genetic testing in cardiomyopathy cases. A genetic test is recommended for diagnosis, prognosis, treatment planning, screening, and reproductive management. Counseling should ideally take place both before and after genetic testing and involve a multidisciplinary team.11
Paldino et al. demonstrated that a genotype-based classification outperformed a phenotype-based classification in predicting key clinical outcomes. This is of particular importance in this heterogeneous group of diseases that have in common the propensity for catastrophic clinical events.6 The 2022 ESC guidelines for managing ventricular arrhythmias and preventing SCD consider specific indications for implanting an ICD in cases with high-risk variants such as those in LMNA, PLN, FLNC, and RBM20, illustrating the significance of genotype-based classification, particularly in assessing the risk of SCD.54
In clinical practice, when a genetic test yields a positive result in a proband, it serves as a valuable tool in aiding the diagnosis of the condition. However, it is essential to recognize that a negative test result does not conclusively rule out the possibility of cardiomyopathy.1
When a proband's genetic test is positive, it should prompt a cascade screening process. This involves testing family members for the presence of the same pathogenic variant identified in the proband with cardiomyopathy. If a relative is found to carry the same pathogenic variant, this triggers a cascade of critical actions. First and foremost, a comprehensive examination is warranted to assess whether the cardiomyopathy phenotype is present in that relative. Should the phenotype be confirmed, a diagnosis of cardiomyopathy can be established, and tailored treatment strategies can be promptly initiated. For relatives who test positive for a pathogenic variant but do not exhibit the clinical phenotype, a proactive approach is essential. These individuals should undergo periodic clinical and diagnostic tests.1
Furthermore, we should highlight the importance of post-mortem molecular testing in cases of cardiac death and suspected cardiomyopathy. This additional genetic information can be invaluable for relatives, aiding in their understanding of potential risks and enabling more informed decisions about their health.
Despite the promise of genetic testing, several critical gaps in our current knowledge persist. Across the cardiomyopathies, the proportion of cases with a confident genetic diagnosis remains relatively low, underscoring the need for ongoing research. For example, in HCM this diagnostic rate can be as low as 40%, and in DCM it is around 30%. The genetic basis of familial DCM remains elusive in a substantial number of cases, and it is also uncertain whether patients with DCM respond differently to pharmacological treatment based on their underlying etiology.1
ConclusionGenetics has gained increasing consideration in recent international guidelines, and the transition toward a genotype-based classification is the direction to pursue. Collaboration among multidisciplinary teams, including experts in cardiomyopathies, arrhythmology, clinical genetics, and pathological anatomy, is imperative to attain this objective.
Conflicts of interestThe authors have no conflicts of interest to declare.
