







Em até um terço dos casos de morte súbita, a autópsia médico‐legal é «branca», estando já contemplada nas recomendações internacionais a possibilidade da realização de autópsia molecular. A importância do teste genético post mortem prende‐se com a identificação de doenças hereditárias, frequentemente doenças com padrão de transmissão autossómico dominante, sendo possível, através de consulta e rastreio de parentes, identificar elementos na família com a doença, não raramente portadores assintomáticos de uma mutação, havendo espaço para alterar o curso de vida dos mesmos. Os autores apresentam três casos clínicos que reforçam o quão importante é o estudo genético post mortem, assim como o estudo familiar e a integração dos dados numa consulta de cardiologia, seja arritmologia, consulta de doença coronária ou miocardiopatias, dependendo da patologia específica, podendo modificar o curso da doença em muitos parentes.
In up to one‐third of cases of sudden death, the medico‐legal autopsy finding is inconclusive, and the option to perform a molecular autopsy is covered in international guidelines. The importance of postmortem genetic testing lies in its ability to identify hereditary diseases, often those with an autosomal dominant transmission pattern, and, through consultations and screening of relatives, to identify family members with a pathogenic mutation, who are often asymptomatic, providing an opportunity to change the course of their lives. The authors present three clinical cases that highlight the importance of postmortem genetic studies and family studies, as well as the integration of the data obtained in a cardiology consultation, which may be for arrhythmology, coronary disease or cardiomyopathy, depending on the specific condition. This could modify the course of the disease in many relatives.
Nos últimos 20 anos a mortalidade cardiovascular tem diminuído, consequentemente as medidas de prevenção de doença coronária e insuficiência cardíaca1. No entanto continuam a ocorrer cerca de 17 milhões de mortes por ano, em todo o mundo, 25% das quais por morte súbita. Define‐se como síndrome de morte súbita arrítmica a morte súbita (morte não traumática e não expectável de indivíduo previamente saudável em 1h após início dos sintomas ou em 24h, após o último contacto com o mesmo) com resultado de autópsia inconclusivo, incluindo avaliação toxicológica e histológica normais1. Em até um terço dos casos de morte súbita, a autópsia médico‐legal é “branca”, estando já contemplada nas recomendações internacionais a possibilidade da realização de autópsia molecular1. A importância do teste genético post mortem prende‐se com a identificação de doenças hereditárias, frequentemente doenças com padrão de transmissão autossómico dominante, sendo possível, através de consulta e rastreio de parentes, identificar elementos na família com a doença, não raramente portadores assintomáticos de uma mutação, havendo espaço para alterar o curso de vida dos mesmos. O estudo genético pode identificar canalopatias em até 35‐50% das causas de morte súbita arrítmica, sendo possível identificar essa mutação patogénica em 25‐35% dos parentes1–4.
Assim, apresentamos três casos clínicos que reforçam o quão importante é o estudo genético post mortem, assim como o estudo familiar e a integração dos dados numa consulta de cardiologia, seja arritmologia, consulta de doença coronária ou miocardiopatias, dependendo da patologia específica, podendo modificar o curso da doença em muitos parentes.
Caso clínico 12010
Doente do género feminino, de 15 anos, com episódios de palpitações, sem síncope. Foram documentados episódios de taquicardia ventricular monomórfica lenta (fig. 1). Realizou ecocardiograma transtorácico, onde foram descritas câmaras cardíacas de dimensões normais e função sistólica biventricular conservada, mas com hipertrabeculação do ventrículo esquerdo. Realizou subsequentemente ressonância magnética cardíaca que diagnosticou ventrículo esquerdo não compactado. A doente realizou estudo eletrofisiológico, onde foi documentada taquicardia ventricular de ramo a ramo, tendo sido decidido não realizar ablação atendendo ao atraso da condução intraventricular em eletrocardiograma (ECG) basal e possibilidade de necessidade de pacing. Foi implantado cardioversor‐desfibrilhador implantável (CDI). Em 2014 foi admitida num serviço de urgência após paragem cardiorrespiratória em ritmo de fibrilhação ventricular com vários choques de CDI, mas acadando a doente por morrer sem viabilidade neurológica.
Caso clínico 2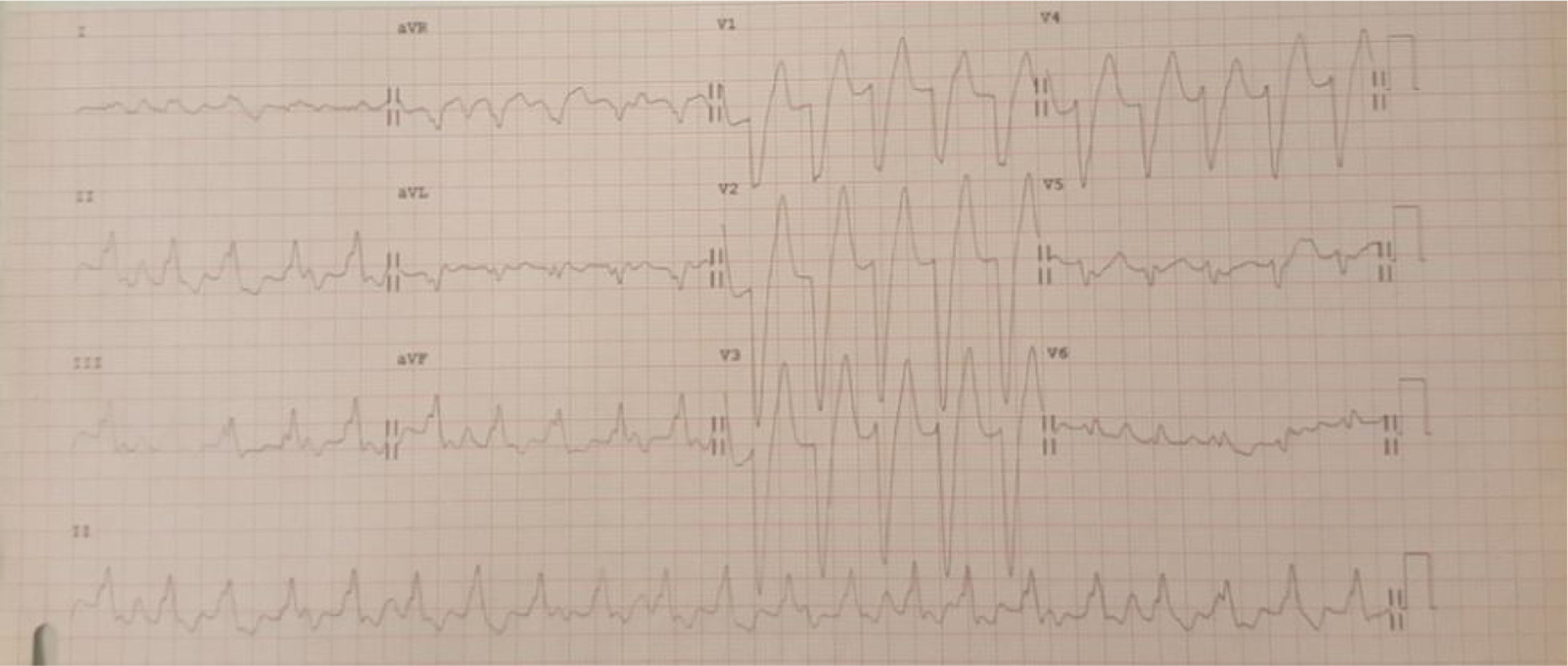
2014
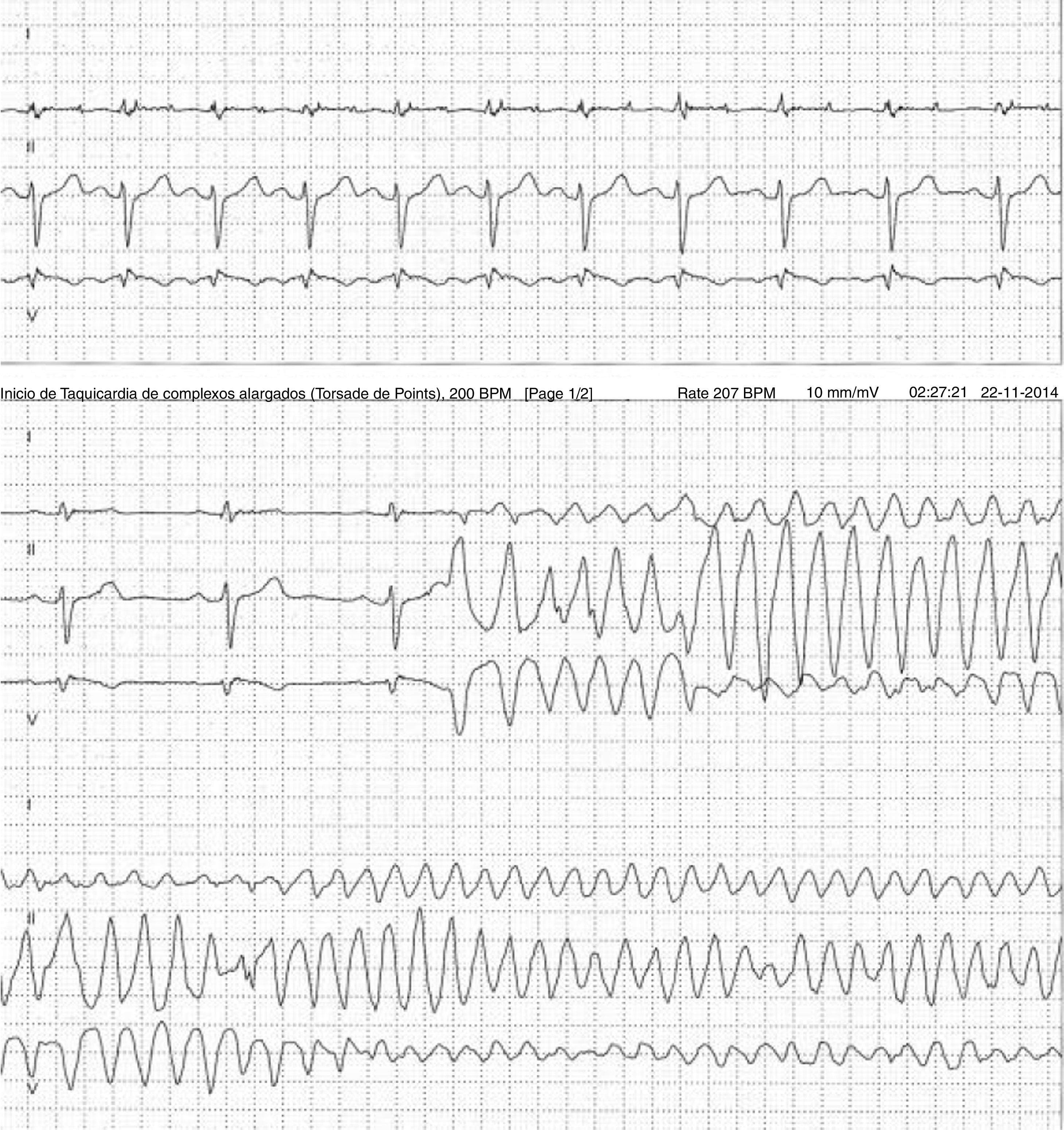
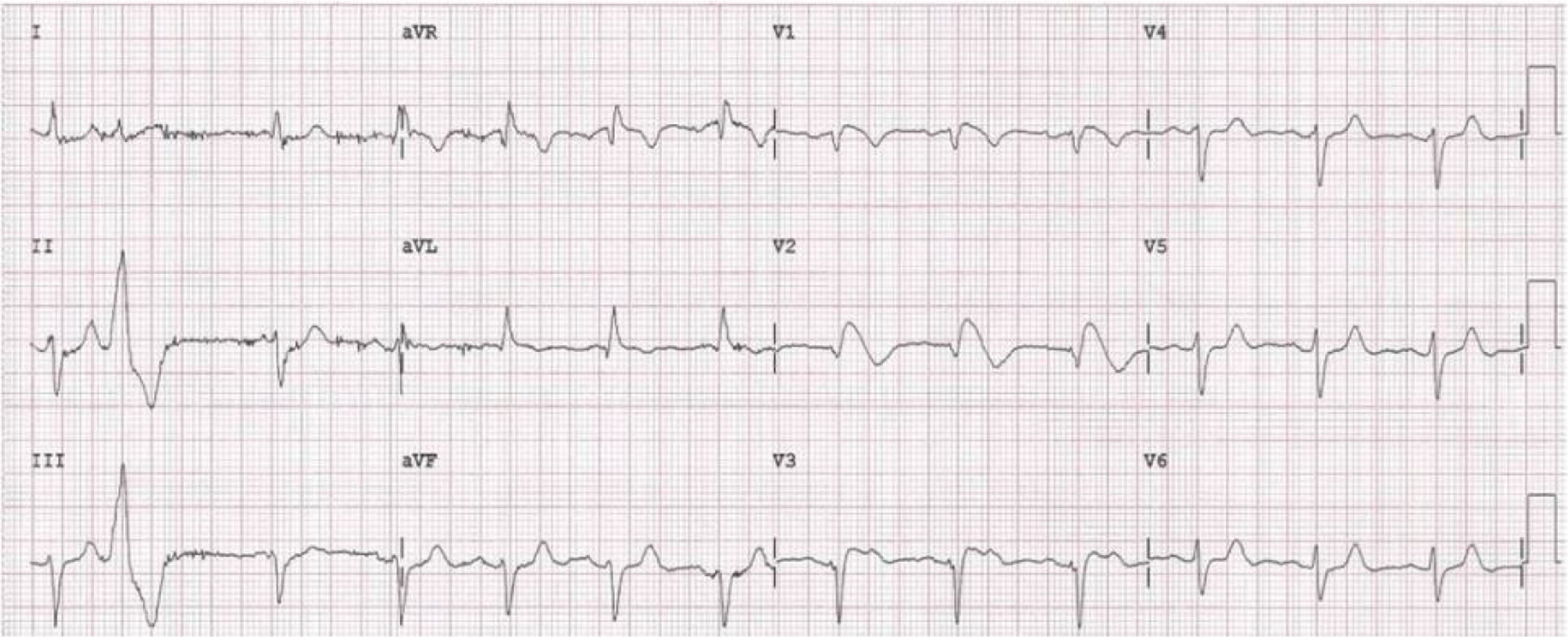
Doente do género feminino, de 59 anos, sem história familiar relevante conhecida pela doente, com episódio de palpitações, sem síncope, realizou ecocardiograma transtorácico que foi normal e Holter de 24h, a pedido de médico de Medicina Geral e Familiar. O Holter de 24h registou taquicardia ventricular polimórfica após extrassístole ventricular R em T (fig. 2), seguida de assistolia e bloqueio auriculoventricular, na totalidade cerca de 4 minutos. Após avaliação do Holter na clínica, a doente foi orientada para o serviço de urgência. Realizou estudo analítico, que foi normal, e eletrocardiograma, com registo de padrão de Brugada tipo 1 (fig. 3). Foi implantado cardioversor‐desfibrilhador implantável. Realizou teste genético, onde foi identificada em heterozigotia a variante 4720G>A no gene no gene SCN5A (MIM 600163), classificada como patogénica. Foi disponibilizada consulta de arritmologia para avaliação de parentes, tendo sido avaliados apenas os dois filhos, ambos do género masculino; segundo a doente, a relação com os outros parentes era má e não estariam interessados na consulta.
2015
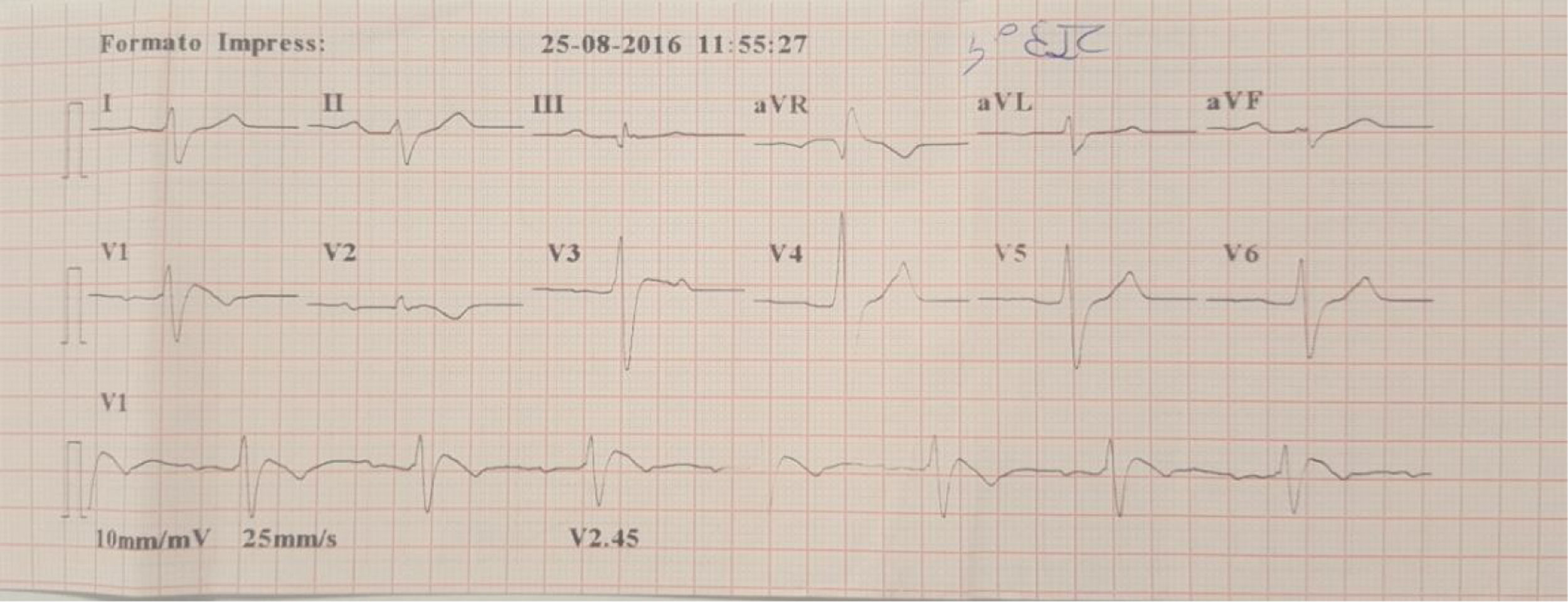
Família referenciada para consulta de cardiologia para avaliação clínica. Trata‐se de parentes de indivíduo do género masculino, de 30 anos, sem história familiar relevante conhecida, assintomático até agosto de 2015, quando, enquanto conduzia veículo pessoal, foi vítima de morte súbita. Foi realizada autópsia médico‐legal sem evidência de causa de morte. Foi requisitado pela equipa do gabinete médico‐legal teste genético no contexto de autópsia molecular. Quando os parentes acederam ao resultado da autópsia, onde foi identificada em heterozigotia a variante 4720G > A no gene SCN5A, foram orientados para consulta de arritmologia para avaliação. Foram avaliados os pais e as três irmãs. Os pais do jovem com morte súbita são assintomáticos, com ECG basal sem alterações. Após consulta de aconselhamento genético, realizaram teste genético, tendo sido identificada a mutação na mãe. Uma das irmãs referia episódios de síncope com convulsões. O ECG, com V1‐V2 no 2°, 3° e 4° espaços intercostais, registava ritmo sinusal, sem alterações. Foi realizado teste de provocação farmacológica com flecainida, com registo de padrão de Brugada tipo1 (fig. 4). Após discussão com a doente, foi decidido implantar cardioversor‐desfibrilhador implantável. Realizou também teste genético, com identificação da mesma mutação identificada nos outros elementos da família. A outra irmã é assintomática, com ECG basal normal, mas portadora da mutação genética. A outra irmã é assintomática, com ECG normal, e não portadora da mutação genética.
Atendendo ao apelido da família e à identificação da mesma mutação do caso clínico 2, e pela agora disponibilidade da família em falar com os restantes parentes, desde logo se concluiu trata‐se da mesma família. Também foi referido que havia uma parente afastada com morte súbita em idade jovem. Após construção da árvore genealógica (fig. 5), verificou‐se que a parente era uma jovem (caso clínico 1) com a história clínica sumária apresentada.
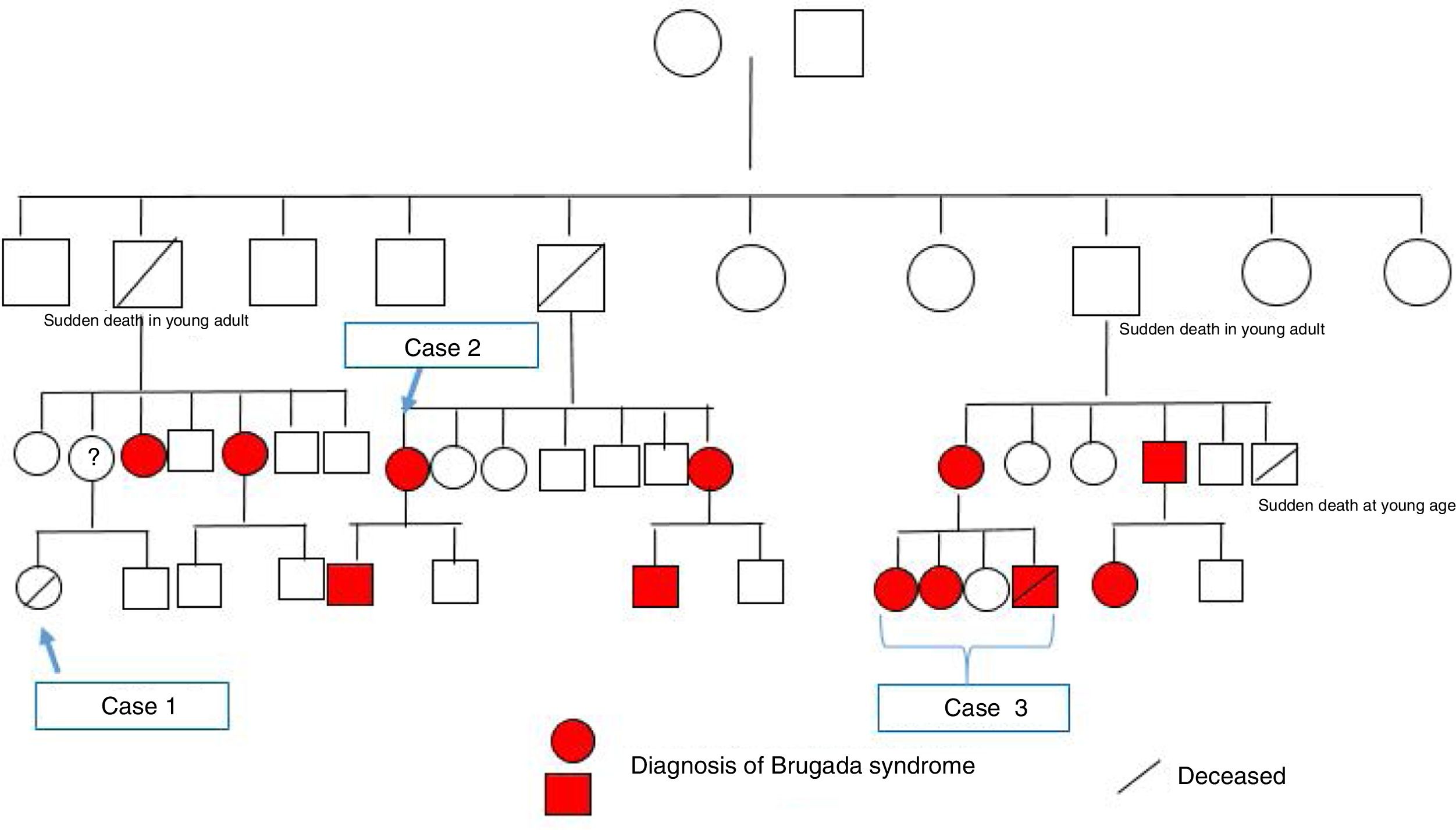
Através da consulta de arritmologia e consulta de genética médica, foi possível a identificação de mais 11 elementos da família portadores da mutação, seis dos quais com padrão de Brugada tipo1 espontâneo, embora seja de realçar que há vários elementos ainda em estudo.
DiscussãoA incidência de morte súbita cardíaca aumenta de forma significativa com a idade, em paralelo com a incidência crescente de doença coronária. Em faixas etárias inferiores a 35 anos é rara, sendo as causas mais prevalentes miocardite, cardiomiopatias, doença coronária em idade precoce, anomalias congénitas das artérias coronárias e canolapatias1,5. Num artigo recente sobre morte súbita em atletas, a síndrome de morte súbita arrítmica foi a principal causa de morte súbita, tendo sido responsável por 56% dos casos em atletas com idade inferior a 18 anos, 44% dos casos dos 18 aos 35 anos e por 26% dos casos acima dos 18 anos6.
A síndrome de Brugada é diagnosticada através de um padrão eletrocardiográfico específico, com elevação do segmento ST ≥ 2mm em uma ou mais derivações precordiais direitas V1 e/ou V2, posicionadas no 2°, 3° ou 4° espaços intercostais, de forma espontânea ou após teste de provocação farmacológica com administração endovenosa de bloqueadores dos canais de sódio, designadamente flecainida, ajmalina, procainamida ou pilsicainida1,7. A importância do diagnóstico prende‐se com o risco de arritmias ventriculares potencialmente fatais inerentes a esta síndrome, assim como com o aconselhamento e rastreio de parentes, atendendo ao padrão de transmissão predominantemente autossómico dominante2.
Esta árvore genealógica impõe algumas questões pertinentes e que devem ser alvo de discussão. Começando pelo caso clínico 3, ao vermos o estudo familiar que se iniciou através da identificação post mortem de mutação associada à síndrome de Brugada, torna‐se inequívoca a importância deste teste nos casos das vítimas de morte súbita sem causa identificada.
Em até um terço dos casos de morte súbita, a autópsia médico‐legal é branca e progressivamente o papel da autópsia genética post mortem vai ganhado mais relevo, estando já contemplada nas recomendações internacionais a possibilidade da sua realização1,2,5,8.
Num estudo em 49 casos de morte súbita de causa não identificada na autópsia, com idade média de 14,2 anos, foi possível identificar mutações genéticas associadas à síndrome de QT longo em cerca de 20% dos casos e em 14% dos casos mutações no gene da rianodina, associada à taquicardia ventricular catecolaminérgica9. Na avaliação retrospetiva dos doentes com mutação nos genes da síndrome de QT longo, 33% dos doentes com morte súbita tinha antecedentes de síncope e 56% história familiar de eventos cardíacos; o estudo genético dos parentes identificou 23 portadores da mutação, com implicações em termos de terapêutica10. Noutro estudo, sobre teste genético post mortem, que inclui 173 doentes com idade média 18,4 ± 12,9 anos (1‐69 anos) com morte súbita e autópsia normal, foram estudadas mutações em genes da síndrome de QT longo e taquicardia ventricular catecolaminérgica, tendo sido identificadas mutações patogénicas em 26% dos doentes9. Ainda noutro estudo que inclui 15 doentes entre um ano e 35 anos, com morte súbita e autópsia não molecular normal, analisou a presença de mutações associadas à síndrome de Brugada, síndrome de QT longo e taquicardia ventricular catecolaminérgica, tendo sido possível identificar mutações patogénicas em 40% dos doentes, e subsequentemente identificação das mesmas mutações em 10 parentes em 1° grau11.
Todos estes estudos reforçam a importância da autópsia molecular em doentes vítimas de morte súbita.
Encontramos na literatura várias indicações da abordagem clínica de parentes de doentes com morte súbita1,2,11,12. Há vários países que têm implantado um protocolo de investigação de doentes com morte súbita e autópsia não molecular normal, assim como protocolos de referenciação e estudo de parentes dos mesmos doentes. Num documento de consenso da Heart Rhythm Society, European Heart Rhythm Association e Asia Pacific Heart Rhythm Society, são explícitas as recomendações sobre a avaliação de parentes de doentes com morte súbita, realçando a importância da história clínica, da eletrocardiografia (eletrocardiograma de 12 derivações com V1‐V2 no 2°. 3° e 4° espaços intercostais, Holter de 24h, prova de esforço e teste de provocação farmacológica) e da imagem (ecocardiografia e eventual ressonância magnética cardíaca)3. Inequivocamente, como uma classe I, está a recomendação do estudo genético dos parentes de vítimas de morte súbita em quem foi identificada uma mutação patogénica, reforçando o valor deste estudo molecular na morte súbita sem identificação de causa.
Em Portugal encontramos alguns constrangimentos do ponto de vista legal, nomeadamente no acesso aos resultados do teste genético post mortem, no âmbito da autópsia médico‐legal; estes testes, requisitados pelos colegas médicos que realizam a autópsia, são enviados de forma sigilosa no relatório final para o Tribunal, sendo o acesso por terceiros a esses resultados apenas possível mediante autorização por parte do Tribunal. Só se a família requisitar, junto do Tribunal, cópia do resultado da autópsia médico‐legal e valorizar os achados encontrados ou entregar ao médico assistente o relatório é que este último terá a possibilidade de valorizar os achados e orientar para consulta hospitalar. Não há, pois, um mecanismo para orientar estes resultados diretamente para uma consulta pré‐estabelecida de avaliação de parentes de doentes com morte súbita.
Outra reflexão sobre esta família é que os parentes não são avaliados em consulta de cardiologia caso o nosso caso índex não os conduza até nós. Verificamos que a doente do caso clínico dois não comunicou aos parentes a possibilidade de avaliação em consulta hospitalar, perdendo‐se uma oportunidade de avaliação/rastreio da doença e identificação de doentes de alto risco de morte súbita.
A última reflexão prende‐se com a doente do caso clínico 1. Nunca poderemos ter a certeza se a doente era portadora da mutação. Também a mãe se recusa a realizar o teste genético, o que, na eventualidade de ser portadora da doença, poderia reforçar a possibilidade da jovem ter uma síndrome de Brugada com uma manifestação atípica da doença. A não compactação do ventrículo esquerdo é uma miocardiopatia com reconhecido sobrediagnóstico13,14. A taquicardia ventricular monomórfica é uma apresentação rara da síndrome de Brugada15–19. Este facto, aliado a uma possibilidade de apresentação rara da síndrome, taquicardia ventricular monomórfica, assim como à ausência de padrão de Brugada tipo1 no ECG basal, pode ter possibilitado o não diagnóstico de síndrome de Brugada nesta doente.
ConclusãoO teste genético post mortem realizado em vítimas de morte súbita com autópsia «branca» demonstra‐se uma mais valia do ponto de vista clínico, permitindo o diagnóstico de doenças hereditárias associadas a arritmias ventriculares malignas e, consequentemente, possibilitando a prevenção da morte súbita. Indo de encontro à situação que já se verifica em outros países, Portugal necessita de criar um protocolo de estudo/rastreio de parentes de vítimas de morte súbita associada a doenças hereditárias.
Conflitos de interesseOs autores declaram não haver conflito de interesses.
