
Nos últimos anos, tem sido crescente o reconhecimento das causas genéticas das doenças cardiovasculares resultado dos significativos progressos das técnicas laboratoriais. Este conhecimento tem permitido a identificação de «novos» fenótipos e a subclassificação das síndromes clínicas, tendo impacto nas decisões terapêuticas e no aconselhamento genético que é facultado às famílias.
No presente documento descreve‐se o «estado da arte» relativamente às principais recomendações para testes genéticos nas doenças cardiovasculares, pretendendo‐se providenciar uma ferramenta útil de consulta para cardiologistas e outros profissionais envolvidos na prestação nos cuidados de saúde a doentes com cardiopatias hereditárias e respetivas famílias.
In recent years, the importance of genetic causes of cardiovascular diseases has been increasingly recognized, as the result of significant advances in molecular diagnosis techniques. This growing knowledge has enabled the identification of new phenotypes and the subclassification of clinical syndromes, impacting the therapeutic approach and genetic counseling offered to affected families.
This paper describes the state of the art of genetic testing in the main cardiovascular diseases, aiming to provide a useful tool to help cardiologists and other health professionals involved in the care of individuals with hereditary heart diseases and their families.
Nos últimos anos, tem sido reconhecida a crescente importância das causas genéticas nas doenças cardiovasculares e os progressos técnicos têm permitido detalhar muitos dos mecanismos moleculares subjacentes.
Os enormes avanços das técnicas de sequenciação genética (next‐generation sequencing – NGS) aumentaram a capacidade de diagnóstico molecular, mas, em muitos casos, à custa da deteção de variantes genéticas de significado (ainda) incerto, que condicionam o aconselhamento genético familiar. Neste contexto, torna‐se fundamental que os cardiologistas e outros profissionais envolvidos na orientação destes doentes/família estejam familiarizados com as indicações, vantagens e limitações dos testes genéticos.
Ao longo deste documento, descrevem‐se as principais indicações para testes genéticos das principais síndromes/doenças cardiovasculares hereditárias e para os testes genéticos post mortem.
Recomendações geraisSempre que apropriado, os médicos devem informar aqueles que os consultam sobre os mecanismos de hereditariedade da doença em causa e quais as implicações para os seus familiares e de os orientar para uma consulta de genética médica.
O aconselhamento genético deve ser sempre efetuado antes e após um teste genético, garantindo que os doentes compreendam todos os benefícios e as limitações dos resultados, sendo que o teste genético só deve ser realizado após o consentimento informado do próprio. A comunicação dos resultados deve ser feita exclusivamente ao próprio doente.
MenoresNo caso de menores de idade, só podem ser pedidos testes genéticos se houver benefício imediato para estes, com o consentimento informado dos seus pais ou tutores, sendo de realçar que não podem ser pedidos testes preditivos para doenças de início habitual na vida adulta, sem prevenção ou cura comprovadamente eficazes. Sempre que adequado o/a menor deverá ser envolvido/a na proporção da sua autonomia.
Testes pré‐sintomáticosConsideram‐se testes pré‐sintomáticos os que permitam a identificação da pessoa como portadora, ainda que assintomática, de alteração genética inequivocamente responsável por dada patologia. Em pessoas saudáveis, o teste pré‐sintomático só pode ser executado no âmbito de uma consulta de genética médica, na sequência de aconselhamento genético, após consentimento informado, escrito. Também neste contexto, os resultados devem ser comunicados ao próprio e não podem ser comunicados a terceiros, incluindo médicos não envolvidos no processo de teste dessa pessoa/família, sem autorização escrita.
Os testes genéticos encontram‐se legislados no Decreto‐Lei 12/2005 de 26 de janeiro, do qual se apresentam os excertos mais relevantes nos suplementos (Supl.1).
Os principais testes utilizados na prática clínica e a lista de genes citados ao longo do documento de abreviaturas utilizadas encontram‐se em suplementos (Supl.2‐4).
Classificação das variantes genéticasAs variantes devem ser classificadas em cinco categorias: patogénicas, provavelmente patogénicas, de significado incerto (VSI), provavelmente benignas, benignas1. De notar que apenas as variantes patogénicas e provavelmente patogénicas devem ser utilizadas para orientar o seguimento diferenciado e disponibilizar estudo de portador a familiares assintomáticos em risco.
Níveis de recomendaçãoA maioria da informação disponível deriva de registos e de estudos não aleatorizados – nível de evidência C. Os níveis de recomendação usados ao longo deste documento são apresentados no material suplementar (Supl.5).
Testes genéticos em familiaresApós a identificação de uma variante genética específica, patogénica ou provavelmente patogénica, num caso índice, é uma indicação classe I2,3, comum a todas as doenças cardíacas hereditárias, a realização de teste genético em familiares, conjuntamente com aconselhamento genético, antes e após o teste genético.
CanalopatiasAs canalopatias são doenças elétricas primárias do coração, que não se acompanham de alterações macroscópicas ou histopatológicas identificáveis pelas metodologias habituais, dado que as alterações funcionais e estruturais se situam a nível molecular, na membrana celular4. São um grupo heterogéneo de patologias nas quais variantes patogénicas nos genes que codificam os canais iónicos originam alterações das correntes iónicas envolvidas no potencial de ação das células cardíacas, conduzindo a arritmias potencialmente fatais5,6.
Síndromes da onda JAs síndromes da onda J referem‐se a situações em que a acentuação da onda/ponto J no ECG está associada a um risco acrescido de arritmias ventriculares7. A síndrome de Brugada (SBr) e a síndrome de repolarização precoce (SRP) são duas manifestações destas síndromes, estando associadas ao desenvolvimento de taquicardia ventricular (TV) polimórfica, fibrilhação ventricular (FV) e, potencialmente, a morte súbita (MS)8. As alterações na onda/ponto J ocorrem em diferentes derivações do ECG – na SBr nas derivações pré‐cordiais direitas, na SRP essencialmente nas inferiores e laterais7,8.
Síndrome de BrugadaDiagnóstico clínicoA SBr é diagnosticada em doentes com padrão eletrocardiográfico de tipo 1 (elevação do segmento ST ≥2mm, em cúpula (coved type), numa ou mais derivações pré‐cordiais direitas, V1 e/ou V2, posicionadas no 2.°, 3.° ou 4.° espaço intercostal), espontaneamente ou após teste de provocação com fármacos bloqueadores dos canais de sódio (como flecainida ou ajmalina)8–10.
Para evitar o sobrediagnóstico, está recomendado que nos casos em que a documentação do padrão eletrocardiográfico de tipo 1 tenha sido obtida apenas após provocação farmacológica, para o diagnóstico de SBr exista adicionalmente pelo menos um dos seguintes critérios: a) FV /TV polimórfica; b) síncope de provável causa arrítmica; c) história familiar de MS antes dos 45 anos na presença de autópsia negativa; d) padrão eletrocardiográfico de tipo 1 em familiares; e) respiração agónica noturna; f) indutibilidade de FV/TV8. Na Tabela S1 (Supl.6) apresenta‐se um score diagnóstico para a SBr8.
Diagnóstico genéticoA SBr tem sido associada a variantes genéticas em múltiplos genes, que codificam principalmente canais de sódio (SCN5A em 11‐28% dos probandos, SCN10A em 5‐17%) e de cálcio (CACNA1C em 7% e CACNB2b em 5%)8.
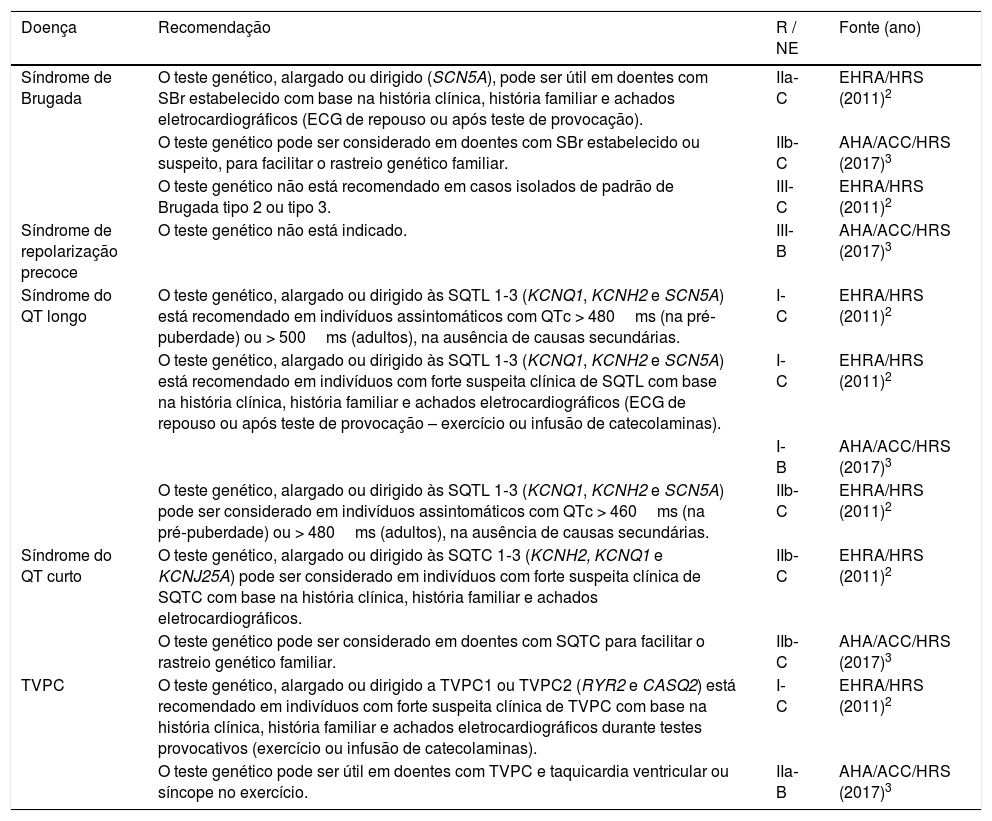
O teste genético não é necessário para o diagnóstico, podendo, contudo, ser útil na confirmação do diagnóstico em doentes com fenótipos duvidosos e em doentes com SBr estabelecida (classe IIb[3] / IIa[2]), particularmente para facilitar o rastreio genético familiar (classe IIb[3]) (Tabela 1).
Recomendações para a realização de testes genéticos nas canalopatias
| Doença | Recomendação | R / NE | Fonte (ano) |
|---|---|---|---|
| Síndrome de Brugada | O teste genético, alargado ou dirigido (SCN5A), pode ser útil em doentes com SBr estabelecido com base na história clínica, história familiar e achados eletrocardiográficos (ECG de repouso ou após teste de provocação). | IIa‐C | EHRA/HRS (2011)2 |
| O teste genético pode ser considerado em doentes com SBr estabelecido ou suspeito, para facilitar o rastreio genético familiar. | IIb‐C | AHA/ACC/HRS (2017)3 | |
| O teste genético não está recomendado em casos isolados de padrão de Brugada tipo 2 ou tipo 3. | III‐C | EHRA/HRS (2011)2 | |
| Síndrome de repolarização precoce | O teste genético não está indicado. | III‐B | AHA/ACC/HRS (2017)3 |
| Síndrome do QT longo | O teste genético, alargado ou dirigido às SQTL 1‐3 (KCNQ1, KCNH2 e SCN5A) está recomendado em indivíduos assintomáticos com QTc > 480ms (na pré‐puberdade) ou > 500ms (adultos), na ausência de causas secundárias. | I‐C | EHRA/HRS (2011)2 |
| O teste genético, alargado ou dirigido às SQTL 1‐3 (KCNQ1, KCNH2 e SCN5A) está recomendado em indivíduos com forte suspeita clínica de SQTL com base na história clínica, história familiar e achados eletrocardiográficos (ECG de repouso ou após teste de provocação – exercício ou infusão de catecolaminas). | I‐C | EHRA/HRS (2011)2 | |
| I‐B | AHA/ACC/HRS (2017)3 | ||
| O teste genético, alargado ou dirigido às SQTL 1‐3 (KCNQ1, KCNH2 e SCN5A) pode ser considerado em indivíduos assintomáticos com QTc > 460ms (na pré‐puberdade) ou > 480ms (adultos), na ausência de causas secundárias. | IIb‐C | EHRA/HRS (2011)2 | |
| Síndrome do QT curto | O teste genético, alargado ou dirigido às SQTC 1‐3 (KCNH2, KCNQ1 e KCNJ25A) pode ser considerado em indivíduos com forte suspeita clínica de SQTC com base na história clínica, história familiar e achados eletrocardiográficos. | IIb‐C | EHRA/HRS (2011)2 |
| O teste genético pode ser considerado em doentes com SQTC para facilitar o rastreio genético familiar. | IIb‐C | AHA/ACC/HRS (2017)3 | |
| TVPC | O teste genético, alargado ou dirigido a TVPC1 ou TVPC2 (RYR2 e CASQ2) está recomendado em indivíduos com forte suspeita clínica de TVPC com base na história clínica, história familiar e achados eletrocardiográficos durante testes provocativos (exercício ou infusão de catecolaminas). | I‐C | EHRA/HRS (2011)2 |
| O teste genético pode ser útil em doentes com TVPC e taquicardia ventricular ou síncope no exercício. | IIa‐B | AHA/ACC/HRS (2017)3 |
ACC – American College of Cardiology; AHA – American Heart Association; ECG – eletrocardiograma; EHRA – European Heart Rhythm Association; HRS – Heart Rhythm Society; NE – nível de evidência; R – nível de recomendação; SBr – Síndrome de Brugada; SQTC – síndrome do QT curto; SQTL – síndrome do QT longo; TVPC – taquicardia ventricular polimórfica catecolaminérgica.
A SRP é diagnosticada em pacientes com padrão de repolarização precoce nas derivações inferiores e/ou laterais e história de MS abortada, FV ou TV polimórfica documentadas8. O padrão de repolarização precoce é definido pela presença de 1) entalhe na porção final do QRS (onda J) ou de empastamento no ramo descendente na onda R, com ou sem elevação do segmento ST; 2) pico da onda J ≥0,1mV em ≥2 derivações contíguas, exceto V1‐3; e 3) duração do QRS (medida em derivações sem entalhe ou empastamento do QRS) <120ms 11. Na Tabela S2 (Supl.6), apresenta‐se um score de diagnóstico da SRP8.
Diagnóstico genéticoA SRP foi associada, até à data, a variantes genéticas em sete genes, que codificam principalmente canais de cálcio (CACNA1C, CACNB2b e CACNA2D1), mas o seu papel etiológico é questionável8 e, consequentemente, o teste genético não está indicado3 (Tabela 1).
Síndromes do QTSíndrome do QT longoDiagnóstico clínicoO diagnóstico da síndrome do QT longo (SQTL) baseia‐se na medição do intervalo QTc, após exclusão de causas secundárias de prolongamento deste intervalo, como fármacos e alterações eletrolíticas10. Para auxiliar o diagnóstico, foi criado um score12 que, para além da duração do QTc, considera outras alterações eletrocardiográficas, sintomas e história familiar (Supl.6, Tabela S3). Assim, na ausência de causas secundárias, o diagnóstico de SQTL é estabelecido se9: 1) QTc ≥480ms em ECGs repetidos (classe I) ou 2) score de risco > 3 (classe I) ou 3) identificação de variante patogénica, independentemente da duração do QTc (classe I) ou 4) QTc ≥460ms em ECGs repetidos e síncope inexplicada (classe IIa).
Diagnóstico genéticoA SQTL está associada a variantes genéticas documentadas em pelo menos 15 genes13. O teste genético identifica variantes patogénicas em aproximadamente 75% dos casos, sendo que três genes são responsáveis por sensivelmente 90% dos testes positivos9 – KCNQ1, KCNH2 e SCN5A (associados às SQTL tipo 1 a 3, respetivamente)9. Nestes casos em particular, o diagnóstico genético reveste‐se de valor prognóstico, dado que os diferentes genótipos associam‐se a maior ou menor risco de morte súbita, principalmente quando associado ao género e à duração do QTc, sendo este risco particularmente elevado em mulheres com SQTL tipo 2 e homens com SQTL tipo 3 e com QTc > 500ms6,14, e dado que os triggers para os eventos arrítmicos são também distintos (na SQTL tipo 1, a atividade física, particularmente a natação; na SQTL tipo 2, ruídos altos e súbitos; na SQTL tipo 3, o repouso ou sono)6,15. Cerca de 20–25% dos doentes com SQTL confirmado geneticamente apresentam QTc com duração normal10,16.
O estudo molecular pode ser dirigido a um gene específico, orientado pelo ECG, fatores precipitantes da síncope ou presença de características sindrómicas. Nos doentes com surdez congénita, cardiopatia congénita, défice cognitivo, perturbação do espetro de autismo e/ou dismorfias, deve ser considerada a hipótese diagnóstica de síndrome de Jervell e Lange‐Nielson, síndrome de Timothy, síndrome de Andersen‐Tawil ou considerar o uso de painéis genéticos, que incluam múltiplos genes relacionados com o SQTL, que permitam o diagnóstico de formas mais raras de SQTL17.
A síndrome de Jervell e Lange‐Nielson é uma síndrome autossómica recessiva ou, menos frequentemente, heterozigótica composta, envolvendo os genes KCNQ1 ou KCNE1, na qual a presença de prolongamento do QTc, habitualmente > 500ms, se associa surdez neurossensorial congénita bilateral profunda e se manifesta habitualmente por síncope em contexto de ativação simpática6,18; a síndrome de Timothy (SQTL tipo 8) associa‐se a variantes patogénicas no gene CACNA1C e caracteriza‐se pela presença adicional de perturbação da condução aurículo ventricular, taquiarritmias, cardiopatias congénitas, dismorfias faciais, das mãos e dos pés e de perturbação do desenvolvimento do espectro do autismo6,19; a síndrome de Andersen‐Tawil (SQTL tipo 7) associa‐se a variantes patogénicas no gene KCNJ2 e, para além do prolongamento do QTC é caracterizada pela presença de onda U, episódios de paralisia muscular periódica hipocaliémica, dismorfias faciais e défice neurocognitivo ligeiro e manifesta‐se habitualmente por palpitações, síncope, ou episódios de paralisia, após repouso prolongado ou após o repouso depois de esforço físico 6,20.
O teste genético, conjuntamente com aconselhamento genético, está indicado em todos os pacientes com o diagnóstico de SQTL ou com forte suspeita clínica (classe I)2,3, podendo ser considerado em indivíduos assintomáticos com QTc prolongado (> 480ms em adultos e > 460ms na pré‐puberdade) na ausência de causas secundárias (classe IIb)2 (Tabela 1).
Síndrome do QT curtoDiagnóstico clínicoO diagnóstico da síndrome do QT curto (SQTC) é estabelecido na presença de QTc ≤340ms (classe I), devendo ser considerado (classe IIa), se QTc ≤360ms e adicionalmente existir: 1) variante genética patogénica confirmada; 2) história familiar de SQTC; 3) MS familiar antes dos 40 anos; ou 4) TV/FV abortada, sem cardiopatia estrutural9.
Diagnóstico genéticoA SQTC está associada a variantes genéticas em três genes que codificam canais de potássio (KCNH2, KNCQ1 e KCNJ2), os quais também se associam à SQTL, mas com alterações da função destes canais em sentidos opostos6,10.
O teste genético pode ser considerado nos indivíduos com SQTC (classe IIb)2,3 para facilitar o rastreio nos familiares de primeiro grau3 (Tabela 1). Ao contrário do que acontece no SQTL, no SQTC o teste genético não tem valor prognóstico.
Taquicardia ventricular polimórfica catecolaminérgicaDiagnóstico clínicoA taquicardia ventricular polimórfica catecolaminérgica (TVPC) é uma síndrome arritmogénica hereditária que tipicamente se manifesta por síncope ou MS adrenergicamente mediadas e secundárias a taquiarritmias ventriculares21. O diagnóstico é estabelecido na presença de: 1) TV bidirecional ou polimórfica induzida pelo exercício ou stress emocional, na presença de coração estruturalmente normal e ECG normal (classe I); ou 2) variante patogénica nos genes RYR2 ou CASQ2 (classe I)9.
Diagnóstico genéticoO teste genético está recomendado nos indivíduos com TVPC (classe IIa(3) / I(2)) (Tabela 1).
MiocardiopatiasAs miocardiopatias são doenças do miocárdio caracterizadas por alterações estruturais e/ou funcionais do músculo cardíaco na ausência de doença coronária, hipertensão, doença valvular ou cardiopatias congénitas, «suficientes» para provocar as alterações observadas22.
Atualmente o estudo molecular é parte integrante da avaliação e orientação de doentes/famílias com miocardiopatias, sendo também considerado um dos critérios de diagnóstico das formas familiares23,24.
Miocardiopatia hipertróficaDiagnóstico clínicoA miocardiopatia hipertrófica (MCH) é definida pela presença de hipertrofia ventricular esquerda inapropriada e desproporcional às condições de carga na ausência de outra condição cardíaca ou sistémica que justifique a magnitude de hipertrofia observada. O critério de diagnóstico consiste na presença de espessura máxima da parede do ventrículo esquerdo ≥15mm em qualquer segmento miocárdico. Em parentes de 1.° grau, é suficiente uma espessura da parede ≥13mm, não explicada de outra forma, em qualquer segmento miocárdico25.
Diagnóstico genéticoO teste genético (dirigido ou com painéis genéticos alargados) está recomendado em doentes com o diagnóstico clínico de MCH (classe I2,25 / classe IIa3,26 / nível A23), principalmente quando se antevê a realização de rastreio genético familiar25,26. O diagnóstico molecular é também recomendado quando a apresentação clínica sugere etiologia genética específica, não sarcomérica (classe I)26.
Em doentes cumprindo os critérios de diagnóstico de MCH, a «positividade» estima‐se entre 30% a 60% dos casos25,27–29, sendo mais elevada nos casos de doença familiar e mais baixa em doentes idosos e em indivíduos com manifestações clínicas não clássicas (início tardio da doença, menor gravidade da hipertrofia, hipertrofia concêntrica ou septal sigmóidea, ausência de eventos adversos)25,28,29.
Em indivíduos com achados clínicos equívocos (como espessura das paredes do ventrículo esquerdo entre 12‐13mm e hipertensão arterial, doença valvular ou prática de desporto concomitantes) o teste genético deve apenas ser realizado após uma avaliação clínica e familiar exaustivas por equipas especializadas (classe IIa)25, uma vez que o resultado pode igualmente ser equívoco: um resultado negativo não exclui o diagnóstico e as VSI são de difícil interpretação25 (Tabela 2).
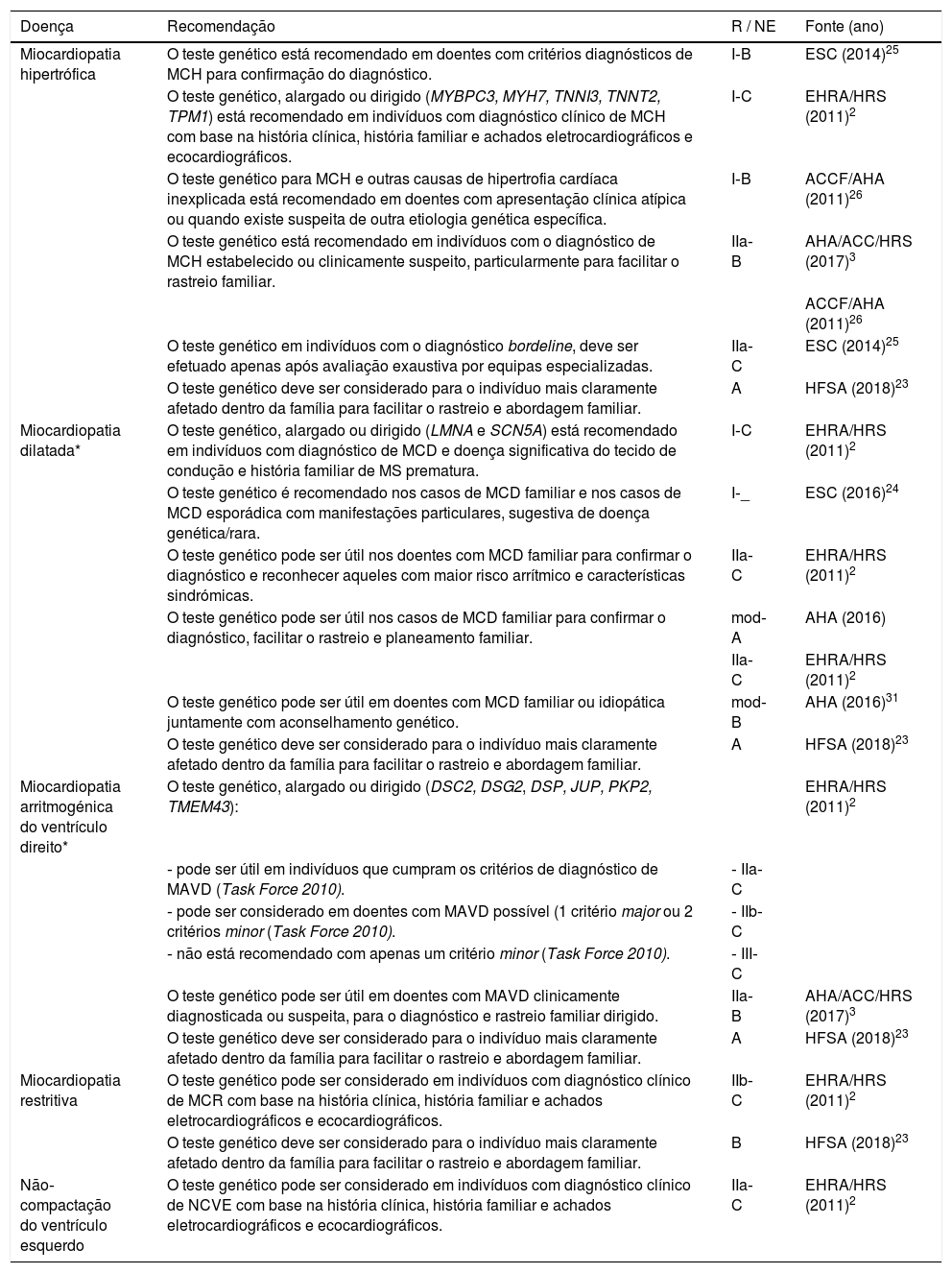
Recomendações para a realização de testes genéticos nas miocardiopatias
| Doença | Recomendação | R / NE | Fonte (ano) |
|---|---|---|---|
| Miocardiopatia hipertrófica | O teste genético está recomendado em doentes com critérios diagnósticos de MCH para confirmação do diagnóstico. | I‐B | ESC (2014)25 |
| O teste genético, alargado ou dirigido (MYBPC3, MYH7, TNNI3, TNNT2, TPM1) está recomendado em indivíduos com diagnóstico clínico de MCH com base na história clínica, história familiar e achados eletrocardiográficos e ecocardiográficos. | I‐C | EHRA/HRS (2011)2 | |
| O teste genético para MCH e outras causas de hipertrofia cardíaca inexplicada está recomendado em doentes com apresentação clínica atípica ou quando existe suspeita de outra etiologia genética específica. | I‐B | ACCF/AHA (2011)26 | |
| O teste genético está recomendado em indivíduos com o diagnóstico de MCH estabelecido ou clinicamente suspeito, particularmente para facilitar o rastreio familiar. | IIa‐B | AHA/ACC/HRS (2017)3 | |
| ACCF/AHA (2011)26 | |||
| O teste genético em indivíduos com o diagnóstico bordeline, deve ser efetuado apenas após avaliação exaustiva por equipas especializadas. | IIa‐C | ESC (2014)25 | |
| O teste genético deve ser considerado para o indivíduo mais claramente afetado dentro da família para facilitar o rastreio e abordagem familiar. | A | HFSA (2018)23 | |
| Miocardiopatia dilatada* | O teste genético, alargado ou dirigido (LMNA e SCN5A) está recomendado em indivíduos com diagnóstico de MCD e doença significativa do tecido de condução e história familiar de MS prematura. | I‐C | EHRA/HRS (2011)2 |
| O teste genético é recomendado nos casos de MCD familiar e nos casos de MCD esporádica com manifestações particulares, sugestiva de doença genética/rara. | I‐_ | ESC (2016)24 | |
| O teste genético pode ser útil nos doentes com MCD familiar para confirmar o diagnóstico e reconhecer aqueles com maior risco arrítmico e características sindrómicas. | IIa‐C | EHRA/HRS (2011)2 | |
| O teste genético pode ser útil nos casos de MCD familiar para confirmar o diagnóstico, facilitar o rastreio e planeamento familiar. | mod‐A | AHA (2016) | |
| IIa‐C | EHRA/HRS (2011)2 | ||
| O teste genético pode ser útil em doentes com MCD familiar ou idiopática juntamente com aconselhamento genético. | mod‐B | AHA (2016)31 | |
| O teste genético deve ser considerado para o indivíduo mais claramente afetado dentro da família para facilitar o rastreio e abordagem familiar. | A | HFSA (2018)23 | |
| Miocardiopatia arritmogénica do ventrículo direito* | O teste genético, alargado ou dirigido (DSC2, DSG2, DSP, JUP, PKP2, TMEM43): | EHRA/HRS (2011)2 | |
| ‐ pode ser útil em indivíduos que cumpram os critérios de diagnóstico de MAVD (Task Force 2010). | ‐ IIa‐C | ||
| ‐ pode ser considerado em doentes com MAVD possível (1 critério major ou 2 critérios minor (Task Force 2010). | ‐ IIb‐C | ||
| ‐ não está recomendado com apenas um critério minor (Task Force 2010). | ‐ III‐C | ||
| O teste genético pode ser útil em doentes com MAVD clinicamente diagnosticada ou suspeita, para o diagnóstico e rastreio familiar dirigido. | IIa‐B | AHA/ACC/HRS (2017)3 | |
| O teste genético deve ser considerado para o indivíduo mais claramente afetado dentro da família para facilitar o rastreio e abordagem familiar. | A | HFSA (2018)23 | |
| Miocardiopatia restritiva | O teste genético pode ser considerado em indivíduos com diagnóstico clínico de MCR com base na história clínica, história familiar e achados eletrocardiográficos e ecocardiográficos. | IIb‐C | EHRA/HRS (2011)2 |
| O teste genético deve ser considerado para o indivíduo mais claramente afetado dentro da família para facilitar o rastreio e abordagem familiar. | B | HFSA (2018)23 | |
| Não‐compactação do ventrículo esquerdo | O teste genético pode ser considerado em indivíduos com diagnóstico clínico de NCVE com base na história clínica, história familiar e achados eletrocardiográficos e ecocardiográficos. | IIa‐C | EHRA/HRS (2011)2 |
ACC – American College of Cardiology; AHA – American Heart Association; EHRA – European Heart Rhythm Association; HFSA – Heart Failure Society of America; HRS – Heart Rhythm Society; MCD – miocardiopatia dilatada; MCH – miocardiopatia hipertrófica; MCR – miocardiopatia restritiva; mod – evidência moderada; MS – morte súbita; NCVE – não‐compactação do ventrículo esquerdo; NE – nível de evidência; R – nível de recomendação.
* Mais recentemente, tem sido reconhecida a diversidade e a sobreposição de fenótipos (entre MCD e MAVD), que partilham o mesmo substrato genético e que se associam a risco arrítmico elevado, originando um novo grupo denominado de “miocardiopatias arritmogénicas” (disfunção ventricular associada a arritmias auriculares, ventriculares ou bloqueio aurículo‐ventricular); nestes casos está também indicado o estudo genético, que deve incluir genes potencialmente arritmogénicos (ex. DSP, LMNA, SCN5A, PLN, TMEM43, FLNC, RBM20, DES).
Doenças hereditárias do metabolismo e outras correspondem a uma pequena, mas importante, fração de doentes genotipados para MCH, sendo as condições mais frequentemente encontradas na população adulta a doença de Anderson‐Fabry, a doença de Danon, a amiloidose e a MCH devida a variantes no gene PRKAG2. O diagnóstico diferencial é crucial, uma vez que estas patologias cursam com história natural e prognóstico muito diferentes e podem implicar atitudes terapêuticas distintas. Nos suplementos (Supl.7) são enumeradas manifestações, cardíacas e extracardíacas, que podem orientar o diagnóstico molecular.
Miocardiopatia dilatadaDiagnóstico clínicoA miocardiopatia dilatada (MCD) é definida pela presença de dilatação e compromisso da função sistólica do ventrículo esquerdo ou de ambos os ventrículos, na ausência de condições de sobrecarga ou de doença coronária «suficientes» para explicar o grau de disfunção. Considera‐se fenotipicamente relacionada a miocardiopatia hipocinética não dilatada (MHND), definida pela presença de disfunção sistólica do ventrículo esquerdo (FEVE < 45%) ou de ambos os ventrículos, na ausência de dilatação ventricular24.
Com a melhoria das técnicas de diagnóstico molecular, tem‐se verificado que 25‐50% dos casos de MCD «idiopática» apresentam uma base genética, predominantemente com transmissão autossómica dominante24,30,31, pelo que é fundamental a avaliação exaustiva da história familiar, envolvendo pelo menos três gerações, nos casos de MCD de novo. Na ausência de uma etiologia genética definitiva, considera‐se que a MCD (ou MHND) é familiar se existirem dois ou mais indivíduos afetados na mesma família ou na presença de um indivíduo com diagnóstico definitivo (MCD ou MHND) e um familiar de 1.° grau com diagnóstico confirmado por autópsia e MS antes dos 50 anos24.
Por outro lado, a ausência de história familiar não exclui etiologia genética e esta deve ser particularmente considerada quando existe perturbação da condução auriculo‐ventricular prévia ou concomitante com a disfunção ventricular ou miopatia esquelética30. Para o diagnóstico da doença em familiares existem ainda outros critérios de diagnóstico (Supl. 8; Tabela S4)24.
Diagnóstico genéticoO teste genético está recomendado (classe I) em doentes com MCD e doença significativa do tecido de condução (bloqueio auriculo‐ventricular de 1.°, 2.° ou 3.° grau) e com história familiar de MS prematura inexplicada2; em doentes com MCD familiar ou nos casos de MCD esporádica associada à presença de manifestações particulares, sugestivas de doença genética/rara24. Nos indivíduos com o diagnóstico de MCD, deve ser testado o indivíduo com o fenótipo mais evidente (nível A)23 (Tabela 2).
Nos suplementos (Supl.9) são enumeradas algumas manifestações sugestivas de doença genética/rara, orientadoras do diagnóstico molecular.
A «positividade» do teste genético aproxima‐se dos 30‐40%31, sendo mais elevada nos casos familiares do que nos casos isolados de MCD (25‐40% versus 10‐25%)23.
Miocardiopatia arritmogénica do ventrículo direitoDiagnóstico clínicoA miocardiopatia arritmogénica do ventrículo direito (MAVD) é definida histologicamente pela substituição progressiva do miocárdio ventricular por tecido fibro‐adiposo, particularmente na região denominada «triângulo da displasia» (entre a câmara de entrada, câmara de saída e ápex do ventrículo direito)22. A MAVD é diagnosticada na presença de disfunção ventricular direita (global ou regional), associada ou não a doença ventricular esquerda, na presença de evidência histológica da doença e/ou alterações do ECG, ecocardiograma ou ressonância magnética cardíaca (RMC) (Supl. 10; Tabela S5)32.
Embora as anomalias estruturais predominem no ventrículo direito, hoje é bem reconhecido que o envolvimento pode ser biventricular e/ou predominantemente do ventrículo esquerdo33. Deve ser elevado o índice de suspeição desta entidade clínica aquando da coexistência de padrões de realce tardio não isquémicos, poupando o subendocárdio na RMC, de anomalias da onda T no ECG e de arritmias ventriculares, particularmente na presença de MS familiar34.
Diagnóstico em casos familiaresApós o diagnóstico de MAVD, o diagnóstico em familiares de 1.° grau, necessita apenas de um dos seguintes critérios: 1) inversão da onda T em V1, V2 e V3 (> 14 anos); 2) potenciais tardios; 3) TV com padrão de bloqueio de ramo esquerdo ou ectopia ventricular frequente (> 200 extrassístoles ventriculares em Holter de 24 horas); 4) ligeira dilatação ou disfunção ventricular direita, global ou segmentar32.
Diagnóstico genéticoO teste genético, dirigido ou alargado, pode ser útil em indivíduos que cumpram os critérios de diagnóstico de MAVD (classe IIa2,3/ nível A23), podendo ser considerado nos casos «possíveis» (classe IIb)2. Na MAVD a «positividade» do teste genético aproxima‐se habitualmente dos 50%35 (Tabela 2).
Miocardiopatia restritivaDiagnóstico clínicoA miocardiopatia restritiva (MCR) é rara e pode ser idiopática, familiar ou secundária a doenças sistémicas. Caracteriza‐se por uma fisiologia restritiva, detetada habitualmente por ecocardiografia, na presença de volumes ventriculares normais (ou diminuídos) e espessura não significativamente aumentada das paredes ventriculares (embora a espessura possa estar aumentada nas doenças infiltrativas)22.
Diagnóstico genéticoO teste genético pode ser considerado em doentes com MCR, depois da ponderação dos achados da anamnese, antecedentes familiares e fenótipo clínico, eletrocardiográfico e ecocardiográfico (classe IIb2/ nível B23) (Tabela 2).
Antes do estudo genético devem ser considerados alguns diagnósticos diferenciais e testes diagnósticos específicos (Supl.11).
Não‐compactação do ventrículo esquerdoDiagnóstico clínicoA não‐compactação do ventrículo esquerdo (NCVE) caracteriza‐se pela presença de trabéculas proeminentes, com recessos intertrabeculares profundos em comunicação com o sangue da cavidade ventricular, sem comunicação com a árvore coronária, sendo possível distinguir uma camada de miocárdio compactado e outra de miocárdio não compactado22. Em alguns indivíduos, a NCVE associa‐se a dilatação ventricular e disfunção sistólica22.
Nem sempre é consensual considerar a NCVE uma miocardiopatia primária e independente ou apenas um traço fenotípico, compartilhado por outras miocardiopatias, outras condições patológicas (doenças neuromusculares, miopatias, doenças mitocondriais) ou fisiológicas (como a gravidez ou prática desportiva)23,36,37.
Para o seu diagnóstico, existem vários critérios baseados nos achados imagiológicos (Supl.12; Tabela S6)38‐44.
A probabilidade de miocardiopatia aumenta quando são cumpridos os critérios diagnóstico quantitativos em eixo curto (de Jenni38 ou de Jacquier43) na presença adicional de um dos seguintes fatores: outro familiar afetado (ou história familiar de miocardiopatia); alterações da contractilidade/compromisso da função ventricular; sintomas/complicações; doença neuromuscular; variante genética potencialmente causal descrita em várias famílias com NCVE45,46.
Diagnóstico genéticoO teste genético pode ser útil em doentes com NCVE, depois da ponderação dos achados da anamnese, antecedentes familiares e fenótipo clínico (particularmente doença neuromuscular), eletrocardiográfico e ecocardiográfico (classe IIa)2. Não está recomendado em indivíduos com NCVE isolada, sem outras alterações na estrutura ou função ventricular, assintomáticos e sem história familiar (Tabela 2).
Rastreio familiar nas miocardiopatiasA avaliação clínica e molecular dos familiares de doentes com miocardiopatias encontra‐se detalhada nos suplementos (Supl.13; Tabela S7).
Aortopatias hereditáriasAs doenças hereditárias da aorta são um grupo heterogéneo de patologias caracterizadas pela ocorrência de aneurismas e/ou disseções num ou mais segmentos da aorta, habitualmente localizados entre o anel aórtico e o nível do diafragma. Dependendo da presença ou ausência de manifestações noutros órgãos, as aortopatias hereditárias podem ser sindrómicas ou não. Os genes identificados codificam maioritariamente proteínas da matriz extracelular, componentes da via TGF‐β ou do aparelho contráctil do músculo liso vascular47,48.
Em doentes com aneurismas/disseções da aorta torácica está recomendado investigar os familiares de 1.° grau de forma a identificar possíveis formas familiares da doença (classe I). Os casos familiares devem ser referenciados a um geneticista para aconselhamento familiar e estudo genético (classe I). Nos casos familiares não sindrómicos, deve ser rastreada a presença de aneurismas também noutros territórios arteriais, incluindo as artérias cerebrais (classe IIa)49.
As principais síndromes associadas a aneurismas/disseções da aorta e as indicações para a avaliação imagiológica da aorta encontram‐se nos suplementos (Supl.14; Tabelas S8.1, S8.2 e S8.3).
Hipercolesterolemia familiarEmbora o conhecimento da hipercolesterolemia familiar (FH) tenha aumentado nas últimas décadas, esta patologia genética comum, potencialmente fatal, mas tratável, permanece subdiagnosticada e subtratada50. É crítico fazer o diagnóstico precoce da FH e instituir medidas terapêuticas adequadas e individualizadas para prevenir a doença aterosclerótica prematura, bem como identificar os parentes afetados e reduzir assim a carga das doenças cardiovasculares nestas famílias.
Diagnóstico clínicoExistem dois sistemas de critérios clínicos para o diagnóstico da FH: os Critérios de Simon Broome Heart Research Trust51 e os Critérios da Clínica de Lípidos da Holanda52. Em Portugal, são habitualmente usados os critérios adaptados de Simon Broome Heart Research Trust:
Hipercolesterolemia familiar possível- (a)
Crianças e jovens menores de 16 anos: Colesterol total > 260mg/dL ou colesterol LDL >155mg/dL*; Adultos: Colesterol total > 290mg/dL ou colesterol LDL >190mg/dL*e
- (b)
História familiar de enfarte do miocárdio antes dos 50 anos em avós e tios ou antes dos 60 anos nos pais, irmãos e filhose/ou
- (c)
História familiar de níveis elevados de colesterol total (> 290mg/dL nos adultos e > 260mg/dL nas crianças e jovens menores de 16 anos) nos pais, irmãos ou filhos; ou colesterol total > 290mg/dL nos avós e/ou tios.
* Valores verificados em duas ocasiões distintas, preferencialmente após 3‐6 meses de implementação de alterações no estilo de vida apropriadas.
Hipercolesterolemia familiar confirmadaIndivíduos que cumpram os critérios acima mencionados e, os próprios ou familiares de 1.° ou 2.° grau (pais, filhos, avós, irmãos, tios), apresentem xantomas tendinosos
ou
Presença de uma variante patogénica/provavelmente patogénica num dos três genes associados a HF: LDLR, APOB, PCSK9.
Diagnóstico genéticoO teste genético da FH deve ser realizado em indivíduos com FH confirmada ou provável, bem como nos familiares em risco, conforme recomendado pelo painel de peritos53. O teste deve incluir os genes LDLR, APOB e PCSK9 e sempre que possível os genes associados a fenocópias da FH: LDLRAP1, APOE, LIPA, ABCG5 e ABCG853. O teste genético fornece informações prognósticas, permitindo efetuar uma estratificação de risco mais refinada.
O rastreio dos familiares em risco (cascata genética) é altamente eficaz na identificação dos indivíduos afetados, que requerem tratamento adequado.
Os indivíduos com FH confirmada devem ser encaminhados para um especialista em FH, particularmente aqueles com FH homozigótica. Todas os indivíduos com FH devem ser avaliados pelo menos anualmente, já que o seu seguimento regular e estruturado pode reduzir a morbilidade e mortalidade por doenças cardiovasculares, através de mudanças de hábitos de vida e medidas terapêuticas precoces e adequadas54.
Cardiopatias congénitasAs cardiopatias congénitas (CC) correspondem a malformações cardiovasculares presentes desde o nascimento e ocorrem em 1‐1,2% dos nados vivos55. Como nem todos os casos são diagnosticados precocemente, a prevalência é difícil de determinar, estando estimada em 13,1/1000 crianças e 6,1/1000 adultos (∼90% são casos esporádicos)56. A maioria das crianças com CC sobrevive até a idade adulta, embora uma proporção significativa necessite de uma ou mais cirurgias ou desenvolva várias complicações, como arritmias ou insuficiência cardíaca.
Os principais fatores etiológicos das CC e a abordagem ao seu diagnóstico genéticos encontram‐se pormenorizados nos suplementos (Supl.15).
Na última década tem sido crescente o número de variantes genéticas associadas a CC, seja formas esporádicas ou hereditárias, sindrómicas ou não. O diagnóstico genético definitivo pode permitir, por exemplo, a identificação de um fenótipo não cardíaco que implique um seguimento e terapêutica particulares e otimizar o aconselhamento genético. No suplemento 15 encontram‐se alguns exemplos de CC sindrómicas frequentes (Tabela S9.1) e dados sobre o risco de recorrência de algumas CC (Tabelas S9.2 e S9.3).
Hipertensão arterial pulmonarDiagnóstico clínicoA hipertensão arterial pulmonar (HAP) define‐se por pressão arterial média ≥ 20mmHg, associada a pressão de encravamento da artéria pulmonar ≤15mmHg e resistências vasculares pulmonares >3 UW, em repouso, avaliadas por cateterismo cardíaco direito57.
Cerca de 70‐80% dos doentes com HAP hereditária autossómica dominante58 e 10‐20% daqueles com HAP esporádica/idiopática59 apresentam variantes no gene BMPR2 (membro da família TGF‐β). No caso de variantes neste gene, a penetrância da doença é maior no sexo feminino60.
Outros genes têm sido associados a HAP, particularmente: TBX4, ATP13A3, GDF2, SOX17, AQP1, ACVRL1, SMAD9, ENG, KCNK3 e CAV158. Variantes no gene EIF2AK4 estão associadas à doença pulmonar veno‐oclusiva/hemangiomatose capilar pulmonar61.
Diagnóstico genéticoO aconselhamento e o diagnóstico genético devem ser oferecidos aos doentes com HAP hereditária ou esporádica/idiopática. Os testes genéticos permitem identificar portadores assintomáticos, mas, dada a penetrância incompleta, não é possível, até à data, predizer quem irá desenvolver a doença. Nestes indivíduos deve ser considerada a vigilância ecocardiográfica59,62. A identificação de variantes no gene EIF2AK4 pode evitar a necessidade de realização de biópsia pulmonar58.
O rastreio pré‐natal na HAP hereditária pode permitir a seleção de embriões por reprodução medicamente assistida63.
Morte súbita cardíaca e testes genéticos post mortem («autópsia molecular»)DefiniçõesA MS é definida como a morte não traumática, inesperada, ocorrendo dentro de uma hora desde o início de sintomas num indivíduo aparentemente saudável. Nos casos não testemunhados, é a morte que ocorre em 24 horas num indivíduo previamente saudável9
O conceito de morte súbita arrítmica ou SADS (Sudden Arrhythmic Death Syndrome) é utilizado quando a causa de morte permanece desconhecida ou incerta após a realização da autópsia, o que ocorre em cerca de 25% a 50% dos casos de MS em jovens64,65.
A morte súbita em crianças com idades inferiores a um ano denomina‐se por síndrome morte súbita infantil. Neste período etário, considera‐se existir uma inter‐relação complexa entre diversos fatores que promovem a ocorrência da MS: o período crítico de desenvolvimento do sistema nervoso autónomo, fatores exógenos (ex. posição no leito), e a vulnerabilidade individual, incluindo fatores genéticos66.
A MS é responsável por 15‐25% da mortalidade na população geral e a sua incidência aumenta significativamente com a idade67–70. Acima dos 40 anos, a doença coronária explica a maioria dos casos71, enquanto nos mais jovens as doenças cardíacas hereditárias, como as miocardiopatias e as canalopatias, são mais frequentes69.
As doenças elétricas primárias (como SQTL, SBr e TVPC) são difíceis de identificar post mortem e é para o seu diagnóstico que é particularmente útil a «autópsia molecular». Na SADS, o teste genético permite a identificação da causa de morte numa percentagem adicional entre 20‐30% dos casos. É importante referir que em doentes previamente diagnosticados com epilepsia, é possível identificar variantes associadas a canalopatias (nomeadamente SQTL e TVPC) em cerca de 20% dos casos72.
Autópsia e testes genéticos post mortemNos casos de MS, a importância do diagnóstico de doenças cardíacas hereditárias reside na possibilidade de identificar familiares vivos em risco (portadores assintomáticos), de forma a intervir precocemente e modificar o curso de vida dos mesmos70.
É indicação classe I a realização de autópsia para investigar a causa de MS9. As recomendações para a realização de autópsias nestes casos foram recentemente atualizadas pela Sociedade Europeia de Patologia Cardiovascular73. Idealmente, deve ser recolhida informação relativa à história médica pessoal e familiar e às circunstâncias da morte. O exame cardíaco deve ser realizado por um patologista experiente e deve ser obtido material biológico para eventual estudo genético. O manuseio deste material requer o consentimento familiar74.
O resultado da autópsia deve ser comunicado à família, devendo existir redes de referenciação que possibilitem a avaliação clínica adequada das famílias. Os testes genéticos post mortem só devem ser realizados após o aconselhamento genético dos familiares75 e estão indicados quando se suspeita de doença cardíaca hereditária, seja pelos achados na autópsia, seja nos casos de SADS (classe IIa)9.
No contexto de SADS, o estudo genético deve incluir genes associados a canalopatias e a miocardiopatias. Apesar das miocardiopatias condicionarem alterações estruturais no miocárdio, estas podem ser muito subtis e não detetadas na autópsia76.
Como nos casos de MS não existe um fenótipo definido a priori, a identificação de uma variante genética não é muitas vezes suficiente para o estabelecimento da sua patogenicidade, daí a importância da integração com os resultados da avaliação familiar72.
Avaliação dos familiaresIndependentemente da realização da autópsia molecular e de acordo com a Sociedade Europeia de Cardiologia, está recomendado o rastreio de doença cardíaca nos familiares de 1.° grau de indivíduos com MS (classe I)9. Essa avaliação deve incluir primeiramente avaliação clínica, ECG, prova de esforço e ecocardiograma e, num segundo nível (dependendo da suspeita clínica), RMC, Holter e prova farmacológica com ajmalina/flecainida9.
Na ausência de um diagnóstico definitivo, depois de uma avaliação sistemática (incluindo ou não o estudo genético), os familiares de 1.° grau devem ser acompanhados de forma periódica e até à idade adulta, altura em que a maioria das doenças já se expressou fenotipicamente9. No entanto, nos casos de autópsias negativas e estudo molecular negativo a taxa de eventos entre os familiares parece ser baixa77.
Conflitos de interesseNada a declarar.
