










Pulmonary arterial hypertension (PAH) is a form of precapillary pulmonary hypertension caused by a complex process of endothelial dysfunction and vascular remodeling. If left untreated, this progressive disease presents with symptoms of incapacitating fatigue causing marked loss of quality of life, eventually culminating in right ventricular failure and death. Patient management is complex and based on accurate diagnosis, risk stratification, and treatment initiation, with close monitoring of response and disease progression. Understanding the underlying pathophysiology has enabled the development of multiple drugs directed at different targets in the pathological chain. Vasodilator therapy has been the mainstay approach for the last few years, significantly improving quality of life, functional status, and survival. Recent advances in therapies targeting dysfunctional pathways beyond endothelial dysfunction may address the fundamental processes underlying the disease, raising the prospect of increasingly effective options for this high-risk group of patients with a historically poor prognosis.
A hipertensão arterial pulmonar (HAP) é um tipo de hipertensão pulmonar pré-capilar causada por um processo complexo de disfunção endotelial e remodelação vascular desfavorável. A história natural desta doença progressiva inclui sintomatologia de insuficiência cardíaca e perda de qualidade de vida, culminando em falência ventricular direita e morte. A gestão dos doentes é complexa, baseando-se num diagnóstico preciso, estratificação de risco e início de terapêutica adequada, sendo essencial a monitoração apertada da resposta e progressão da doença.
A compreensão da fisiopatologia subjacente permitiu o desenvolvimento de múltiplos fármacos dirigidos a diferentes alvos na cadeia patológica. A terapêutica vasodilatadora destaca-se como a abordagem de base nas últimas décadas, tendo permitido melhoria significativa da qualidade de vida, estado funcional e sobrevida dos doentes. Os avanços terapêuticos recentes, com o advento da utilização de fármacos dirigido aos mecanismos subjacentes à doença, trazem a promessa de opções terapêuticas cada vez mais eficazes para este grupo de doentes de alto risco, com um prognóstico historicamente reservado.
Pulmonary arterial hypertension (PAH) is a condition characterized by a complex process of vascular dysfunction, smooth muscle cell, and remodeling of small pulmonary vessels, leading to increased pulmonary vascular resistance (PVR) and pulmonary artery (PA) pressure.
In the latest (2022) European Society of Cardiology/European Respiratory Society (ESC/ERS) guidelines for the diagnosis and treatment of pulmonary hypertension (PH),1 PAH is classified as group 1 PH, comprising subgroups of conditions with similar pathophysiological mechanisms, clinical presentation, hemodynamic characteristics, and therapeutic management. The subgroups are listed in Table 1.
Pulmonary arterial hypertension subtypes.
| 1.1 | Idiopathic |
| 1.1.1 | Non-responders at vasoreactivity testing |
| 1.1.2 | Acute responders at vasoreactivity testing |
| 1.2 | Heritable |
| 1.3 | Associated with drugs and toxins |
| 1.4 | Associated with: |
| 1.4.1 | Connective tissue disease |
| 1.4.2 | HIV infection |
| 1.4.3 | Porto-systemic shunts |
| 1.4.4 | Congenital heart disease |
| 1.4.5 | Schistosomiasis |
| 1.5 | PAH with features of venous/capillary (PVOD/PCH) involvement |
| 1.6 | Persistent PH of the newborn |
PAH: pulmonary arterial hypertension; PCH: pulmonary capillary hemangiomatosis; PH: pulmonary hypertension; PVOD: pulmonary veno-occlusive disease.
Hemodynamic parameters are key in defining and classifying PH. By definition, PAH, similar to chronic thromboembolic PH, is classified as pre-capillary PH as both primarily affect the pre-capillary pulmonary vasculature. The absolute threshold for defining PH was first set at a mean PA pressure (mPAP) ≥25 mmHg in a somewhat arbitrary fashion in the first World Symposium for PH (1973). Since then, real-world evidence provided by large retrospective studies and registries has led to a revision of this figure. The latest ESC/ERS guidelines define PH as mPAP >20 mmHg at rest, defining group 1 PAH as having both PVR <2 Wood units (WU) and PA wedge pressure (PAWP) <15 mmHg, thus excluding a predominant postcapillary component. This distinction in essential, considering the different therapeutic approaches for these subtypes.
PAH is a progressive disease, presenting with symptoms of incapacitating fatigue causing marked loss of quality of life, eventually culminating in right ventricular (RV) failure and death.
Targeted therapies have been developed in recent decades, effectively changing the disease's natural history and improving prognosis. Nevertheless, it remains a challenging condition, with significant morbidity and mortality. Over the last few years, new targeted therapies have appeared based on a deeper understanding of the pathophysiology underlying PAH.
In a rapidly evolving field, this article aims to review current disease management, focusing on pathophysiology, risk stratification, and therapeutic approaches according to the latest guidelines and most recent clinical trials.
EpidemiologyThe annual incidence of PAH in Europe is estimated to be approximately six cases per million individuals per year, with a prevalence of about 50 cases per million.2 It predominantly affects young women, particularly in the case of heritable PAH, with a female-to-male ratio of 2:1. Mean age at diagnosis ranges between 30 and 60 years.3
Mutations in the gene encoding bone morphogenetic protein receptor type 2 (BMPR2), a member of the transforming growth factor beta (TGF-β) superfamily, have been identified as the most frequent cause of heritable PAH.4 In asymptomatic carriers of a BMPR2 mutation, the annual incidence of PAH is 2.3%, and females have 3–4 times higher risk.4
Several other conditions are associated with PAH. Schistosomiasis is thought to be the leading cause of PAH globally, owing to its high prevalence in low- and middle-income countries; it is estimated to affect more than 230 million people worldwide, of whom 10% develop hepatosplenic disease, 5% of whom may develop PAH.5
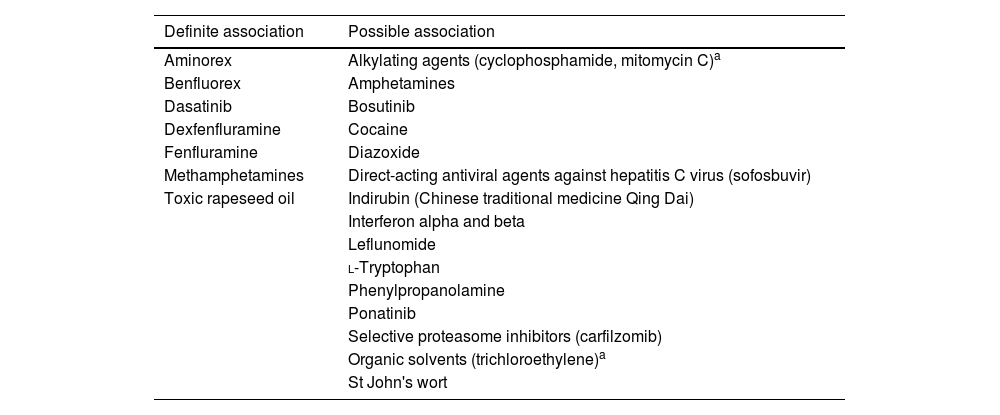
The prevalence of PAH in patients with connective tissue disease is considerable, ranging from around 7% in patients with systemic sclerosis (SSc)6 to 0.5–17% in systemic lupus erythematosus.7 Around 1% of HIV carriers may develop PAH. Congenital heart disease and portal hypertension are also important risk factors for developing PAH.8 Its prevalence does not appear to be influenced by the severity of portal hypertension or liver disease, as it occurs in 2–6% of patients with portal hypertension. Additionally, several drugs and toxins are associated with the development of PAH (Table 2).
Drugs and toxins associated with pulmonary arterial hypertension.
| Definite association | Possible association |
|---|---|
| Aminorex | Alkylating agents (cyclophosphamide, mitomycin C)a |
| Benfluorex | Amphetamines |
| Dasatinib | Bosutinib |
| Dexfenfluramine | Cocaine |
| Fenfluramine | Diazoxide |
| Methamphetamines | Direct-acting antiviral agents against hepatitis C virus (sofosbuvir) |
| Toxic rapeseed oil | Indirubin (Chinese traditional medicine Qing Dai) |
| Interferon alpha and beta | |
| Leflunomide | |
| l-Tryptophan | |
| Phenylpropanolamine | |
| Ponatinib | |
| Selective proteasome inhibitors (carfilzomib) | |
| Organic solvents (trichloroethylene)a | |
| St John's wort |
PAH is the result of a complex process involving multiple cellular signaling pathways, which not only develop and work in parallel but also feed one another, perpetuating the vicious cycle underlying pulmonary vascular remodeling (Figure 1).

Pathophysiology and current therapeutic targets of pulmonary arterial hypertension. cAMP: cyclic adenosine monophosphate; cGTP: cyclic guanosine triphosphate; DLCO: diffusion capacity of the lung for carbon monoxide; ERA: endothelin receptor antagonist; GMP: guanosine monophosphate; GTP: guanosine triphosphate; NO: nitric oxide; PCA: prostacyclin analog; PDE-5: phosphodiesterase 5; PGI2: prostacyclin; PRA: prostacyclin receptor agonist; sGC: soluble guanylate cyclase.
Three main pathologic processes can be identified: vasoconstriction, thrombosis, and remodeling.9 Histopathologic analysis of biopsy tissue and explanted lungs has lead to a deeper understanding of the disease process (Figure 2).
 Muscular hypertrophy of the media and proliferation of intimal cells narrow the lumen of a pulmonary artery branch; adventitial fibrosis can also be detected; (b) an artery shows concentric laminar fibrosis of the intima, imparting an onion skin-like appearance to the vessel profile; (c) a pulmonary artery lumen is fully occluded; (d) the wall of a small artery is replaced by fibrin and inflammatory cells, causing loss of its architecture, in this example of necrotizing vasculitis; (e–g) a glomeruloid network of small vascular channels, the hallmark of plexiform lesions, can be seen arising from muscular arteries; (h) angiomatoid lesions, characterized by dilated and convoluted vessels with a thin wall, can be seen distal to a plexiform lesion (right corner).")
Histopathology of vascular changes of pulmonary hypertension in explanted lungs. (a) Muscular hypertrophy of the media and proliferation of intimal cells narrow the lumen of a pulmonary artery branch; adventitial fibrosis can also be detected; (b) an artery shows concentric laminar fibrosis of the intima, imparting an onion skin-like appearance to the vessel profile; (c) a pulmonary artery lumen is fully occluded; (d) the wall of a small artery is replaced by fibrin and inflammatory cells, causing loss of its architecture, in this example of necrotizing vasculitis; (e–g) a glomeruloid network of small vascular channels, the hallmark of plexiform lesions, can be seen arising from muscular arteries; (h) angiomatoid lesions, characterized by dilated and convoluted vessels with a thin wall, can be seen distal to a plexiform lesion (right corner).
Constriction of pulmonary arterioles stems from an imbalance in the production and secretion of vasodilators and antiproliferative cytokines, such as prostacyclin (PGI2) and nitric oxide (NO),10 and proproliferative and vasoconstrictive factors such as endothelin (ET).11
Disruption of the equilibrium between PGI2 and thromboxane A2 (TXA2), associated with endothelial dysfunction secondary to hypoxia and inflammatory insults, further potentiates the vasoconstrictive environment that characterizes the PAH phenotype.12
Patients with a phenotype driven mainly by vasoconstriction, without the presence of significant vascular remodeling or thrombotic events, are in an early and potentially reversible stage of the disease and are frequently responders to vasodilator therapy (see the section “Currently approved therapies”).
ThrombosisThrombotic events have long been hypothesized to play an important role in PAH. Coagulopathies such as protein C and S deficiency, as well as increased activity of von Willebrand factor, have been reported in patients with PAH.13,14 The association of cytokine imbalance and inflammation with a hypercoagulable state supports the theoretical role of thrombosis.
Warfarin use had a dominant position in patient management for years.15 However, study results are controversial, as they stem mostly from small single-center studies, and there is a lack of placebo-controlled randomized trials. More recent evidence has been obtained through the analysis of large PAH registries, particularly COMPERA16 and REVEAL.17
The lack of clear positive results associated with anticoagulants, together with evidence of increased risk for death in scleroderma-associated PAH, has eroded confidence in the systematic use of these drugs in PAH.17 The current ESC guidelines18 make no general recommendation, and state that individual decision-making is required.
Vascular remodelingVascular remodeling is a cornerstone of disease progression, driven by PA endothelial cell (PAEC) dysfunction and PA smooth muscle cell (PASMC) and fibroblast proliferation, activation, and migration.19 The result is vascular wall remodeling, consisting of intimal proliferation and medial and adventitial layer hypertrophy, as well as extracellular matrix deposition,20 culminating in increased PVR. Smooth muscle cell migration leads to muscularization of otherwise non-muscularized distal capillaries.21
PAEC dysfunction has long been regarded as a major factor in the disease's pathogenesis.22 As described above with vasoconstriction, imbalance between anti- and pro-mitogenic factors drives the proliferative, tumor-like anti-apoptotic phenotype.9 A reduction in NO levels also leads to dysregulation of smooth muscle cell proliferation and migration and overproduction of collagen products, and favors in-situ platelet thrombogenicity.21
Plexiform lesions may develop; these glomeruloid-like structures consist of a network of vascular channels lined with endothelial cells and a core of specialized and apoptosis-resistant myofibroblasts, smooth muscle cells, or even undifferentiated mesenchymal cells.23
Mitochondrial dysfunction also plays a role, with a metabolic shift similar to that of cancer cells (the Warburg effect) stimulating the proliferation of vascular fibroblasts.24
DiagnosisHemodynamics is the basis for PH characterization; however, diagnosis relies on exploration of the overall clinical context and findings from complex multisystemic investigations.
The first step in diagnosing PAH is clinical suspicion of PH. Unexplained fatigue, albeit highly nonspecific, is often the initial manifestation. Throughout disease progression, symptoms will appear with progressively less exertion. Other symptoms may be present and point toward the primary etiology. Physical examination may also reveal signs that aid in identifying the underlying cause of PH (Figures 3–5). Cardiac auscultation findings may strengthen suspicion with specific findings (Figure 6). Signs indicating RV failure tend to appear in the later stages of PAH, necessitating prompt medical intervention.
. WHO-FC: World Health Organization functional class.")

Left: clinical signs suggesting right ventricular failure; right: signs suggesting possible underlying etiologies of pulmonary arterial hypertension. CHD: congenital heart disease; CTEPH: chronic thromboembolic pulmonary hypertension; GERD: gastroesophageal reflux disease; HHT: hereditary hemorrhagic telangiectasia; PDA: patent ductus arteriosus; PPHTN: portopulmonary hypertension; PVOD: pulmonary veno-occlusive disease; RV: right ventricular; SSc: systemic sclerosis.


An initial assessment should include a comprehensive medical and family history, a thorough physical examination, blood tests, and a resting electrocardiogram. Electrocardiographic findings associated with PH include P pulmonale (tall, peaked P waves), right bundle branch block, and voltage criteria for right ventricular hypertrophy.25
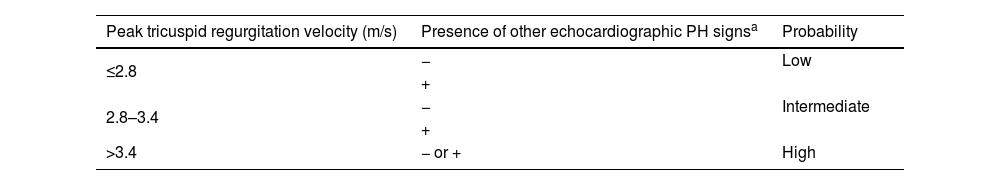
The second step is detecting a high likelihood of PH, through noninvasive lung and cardiac testing. Echocardiography plays a crucial role in the assessment of patients with suspected PH, offering valuable insights into both the probability of PH and potential underlying cardiac abnormalities. This noninvasive imaging modality is a cornerstone of the initial assessment, aiding in the identification of key hemodynamic parameters and structural changes indicative of PH. Direct measurements such as peak tricuspid regurgitation velocity, together with other indirect parameters, enable the probability of PH to be determined (Table 3). The size and respirophasic variation of the inferior vena cava, right atrial area, PA diameter, and RV function parameters, including the tricuspid annular plane systolic excursion (TAPSE)/pulmonary artery systolic pressure (PASP) ratio, offer valuable information regarding the severity and progression of PAH. Moreover, echocardiography serves as a comprehensive tool for identifying underlying cardiac disorders that may contribute to PH. For instance, the presence of congenital heart disease such as atrial or ventricular septal defect can lead to pulmonary vascular remodeling and subsequent PH. Similarly, left heart disease, including valvular abnormalities or ventricular dysfunction, may result in elevated pulmonary pressures due to increased pulmonary venous congestion, indicating a postcapillary component.
Echocardiographic probability of pulmonary hypertension.
| Peak tricuspid regurgitation velocity (m/s) | Presence of other echocardiographic PH signsa | Probability |
|---|---|---|
| ≤2.8 | − | Low |
| + | ||
| 2.8–3.4 | − | Intermediate |
| + | ||
| >3.4 | − or + | High |
PH: pulmonary hypertension.
Chest radiograph findings such as enlargement of the right heart and pulmonary artery silhouette are consistent with PAH, but their presence is not ubiquitous.
Pulmonary function testing (PFT), with forced spirometry, plethysmography, diffusion capacity of the lung for carbon monoxide (DLCO), and arterial blood gas analysis is essential in identifying underlying pulmonary disease. In PAH, PFT is usually normal or may show mild abnormalities,26 whereas more severe abnormalities can be found in patients with PAH associated with congenital heart disease and PH associated with lung disease.27 DLCO is typically slightly reduced; severe reduction (<45% of predicted value) may suggest the presence of venous/capillary involvement (pulmonary veno-occlusive disease [PVOD] or pulmonary capillary hemangiomatosis [PCH]). Risk factors like scleroderma or exposure to organic solvents, or group 3 PH associated with emphysema or interstitial lung disease, should be considered.
PaO2 is generally normal or slightly reduced.28 Severe reduction may indicate structural heart disease such as a patent foramen ovale, or other abnormalities with right-to-left shunting.3
Cardiopulmonary exercise testing (CPET) is valuable in assessing the mechanisms behind exercise intolerance, especially in patients with multiple comorbidities.18 The typical pattern observed in PH patients includes low end-tidal partial pressure of carbon dioxide (PETCO2), high ventilatory equivalent for carbon dioxide (VE/VCO2), low oxygen pulse (VO2/heart rate), and low peak oxygen uptake (VO2).29 Normal peak VO2 indicates a low likelihood of PAH in populations at risk, such as SSc patients.30
Computed tomography pulmonary angiography (CTPA) is a valuable exam for assessing these patients, providing information on the lung parenchyma and vascular dilation, detecting signs of PVOD, and excluding thrombotic phenomena or major congenital heart defects.
Once the diagnosis of PH is strongly suspected, patients should be referred to PH reference centers for optimal management.
The third step, confirmation, relies solely on right heart catheterization (RHC). This invasive test is the gold standard method to confirm the diagnosis of PH and provides a comprehensive assessment of pulmonary hemodynamics.31 The results of this procedure can vary widely depending on operator experience and patient volume status.32
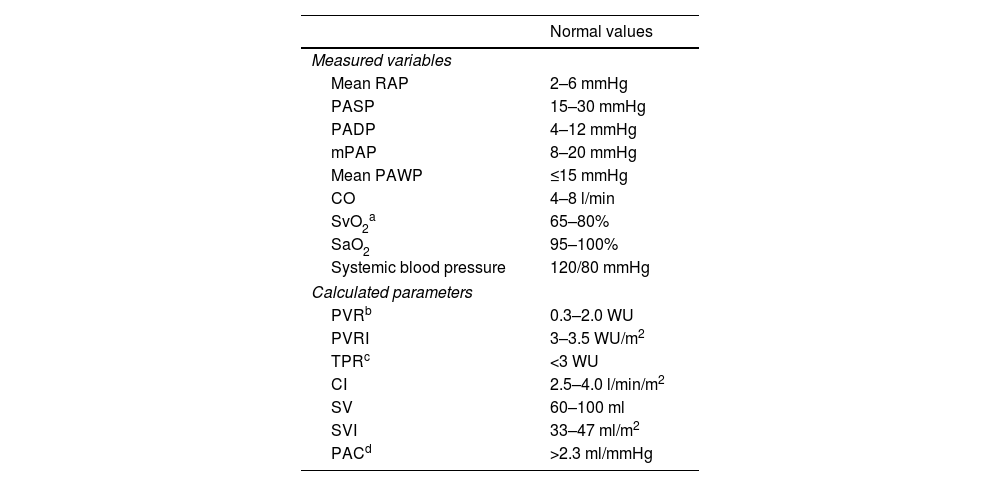
Routinely measured hemodynamic parameters are presented in Table 4.
Hemodynamic measures obtained during right heart catheterization.
| Normal values | |
|---|---|
| Measured variables | |
| Mean RAP | 2–6 mmHg |
| PASP | 15–30 mmHg |
| PADP | 4–12 mmHg |
| mPAP | 8–20 mmHg |
| Mean PAWP | ≤15 mmHg |
| CO | 4–8 l/min |
| SvO2a | 65–80% |
| SaO2 | 95–100% |
| Systemic blood pressure | 120/80 mmHg |
| Calculated parameters | |
| PVRb | 0.3–2.0 WU |
| PVRI | 3–3.5 WU/m2 |
| TPRc | <3 WU |
| CI | 2.5–4.0 l/min/m2 |
| SV | 60–100 ml |
| SVI | 33–47 ml/m2 |
| PACd | >2.3 ml/mmHg |
CI: cardiac index; CO: cardiac output; mPAP: mean pulmonary artery pressure; PAC: pulmonary arterial compliance; PADP: pulmonary artery diastolic pressure; PASP: pulmonary artery systolic pressure; PAWP: pulmonary artery wedge pressure; PVR: pulmonary vascular resistance; PVRI: pulmonary vascular resistance index; RAP: right atrial pressure; SaO2: arterial oxygen saturation; SV: stroke volume; SVI: stroke volume index; SvO2: mixed venous oxygen saturation; TPR: total pulmonary resistance; WU: Wood units.
As previously stated, PH is defined by mPAP >20 mmHg. A precapillary PH phenotype is defined by mPAP >20 mmHg, PAWP ≤15 mmHg, and PVR >2 WU. In patients with borderline PAWP, fluid bolus or exercise hemodynamics may reveal ventricular diastolic dysfunction and a postcapillary component.
The concept of ‘exercise PH’ has been redefined as an mPAP/CO slope >3.0 mmHg/L/min, bearing in mind that the previous definition of mPAP >30 mmHg during exercise (abandoned in 2008) included a significant number of healthy athletes with high CO that was not necessarily pathologic.3
Another indicator recommended for distinguishing between pre- and postcapillary PH at exercise is a PAWP/CO slope with a threshold of >2 mmHg/L/min. This may be particularly important in cases of borderline PAWP between 13 and 15 mmHg, which poses a diagnostic challenge between PAH and PH associated with left heart disease. When PAWP falls within this range, it suggests the possibility of a mixed or indeterminate etiology of PH, in which both pre- and postcapillary factors may contribute to elevated pulmonary pressure. In such cases, additional assessments, including clinical history, physical examination, imaging studies, and hemodynamic response to exercise or other interventions, are necessary to determine the underlying pathology and guide treatment decisions.
Despite its widespread acceptance, further standardization of RHC is necessary to ensure its optimal use in routine clinical practice. To ensure accurate assessment of directly measured or calculated hemodynamic parameters, attention should be directed toward standardizing procedures, such as the position of the pressure transducer and the volume of catheter balloon inflation. Measurement of PAWP is susceptible to over- or under-wedging, potentially leading to false readings. Consequently, errors in RHC measurement and data interpretation can complicate differentiation of PAH from other PH disorders, resulting in misdiagnosis. Beyond diagnosis, the role of RHC, in conjunction with noninvasive tests, is rapidly expanding to include monitoring treatment response and establishing the prognosis of patients diagnosed with PAH.
The fourth step, classification, is of the utmost importance to properly identify the underlying cause of PH and to direct therapy.
An extensive clinical history is crucial and should cover all the clinical subgroups (Table 5).
Important data from clinical history that may elucidate the etiology of pulmonary hypertension and assist with an appropriate clinical classification.
| Relevant details from medical history | Clinical classification groups/subgroups |
|---|---|
| Female gender | Subgroup 1.1.1 |
| Long-term onset of symptoms | |
| Dissociated episodes of (pre-) syncope in WHO functional class II | |
| Family history of PH or death from unknown cause at a young age | Subgroup 1.2 |
| Consumption of or exposure to drugs and toxins specified in Table 2 | Subgroup 1.3 |
| Febrile syndrome | Subgroup 1.4.1 |
| Arthralgias | |
| Myalgias | |
| Dysphagia | |
| Pyrosis | |
| Raynaud's phenomenon | |
| Digital ulcers | |
| Skin changes (e.g. thickening and tightening of the skin on the hands, telangiectasias) | |
| Alopecia | |
| Known HIV infection | Subgroup 1.4.2 |
| Risk factors for HIV infection (e.g., multiple sexual partners, transfusion of blood products, intravenous drug use) | |
| Heavy alcohol consumption | Subgroup 1.4.3 |
| Jaundice | |
| Pruritus | |
| Increase in abdominal volume | |
| Family history of liver disease, known infection with hepatotropic viruses, risk factors for infection with hepatotropic viruses (e.g. transfusion of blood products) | |
| Known cardiac shunting | Subgroup 1.4.4 |
| Past cardiac surgery | |
| Past percutaneous cardiac intervention | |
| Digital clubbing | |
| Hemoptysis | |
| Travel or origin from places with at least a moderate prevalence of schistosomiasis (e.g. sub-Saharan Africa, north-eastern Brazil, near the Yangtze River in China, and in southeast Asia and the Middle East) | Subgroup 1.4.5 |
| Male gender | Subgroup 1.5 |
| Older age | |
| Heavy smoking | |
| Exposure to organic solvents (e.g. trichloroethylene) or alkylating agents | |
| Rapid progression of symptoms | |
| History of intolerance to specific pulmonary vasodilator drugs | |
| Risk factors for atherosclerotic disease | Group 2 |
| Coronary disease, valvular disease, or cardiomyopathy | |
| Family history of sudden cardiac death | |
| Symptoms suggestive of pulmonary congestion (orthopnea or paroxysmal nocturnal dyspnea) | |
| Smoking | Group 3 |
| Digital clubbing | |
| Exposure to air pollution from biomass combustion | |
| Environmental/occupational exposure to agents with known association with lung disease (e.g. asbestos, silica, beryllium, bird feathers, mold) | |
| Prolonged exposure to altitudes >2500 m | |
| Productive cough | |
| Hemoptysis | |
| History of PE | Group 4 |
| Permanent intravascular devices | |
| Inflammatory bowel diseases | |
| Essential thrombocythemia | |
| Splenectomy | |
| High-dose thyroid hormone replacement | |
| Malignancy | |
| Inherited and acquired chronic hemolytic anemia or chronic myeloproliferative disorders | Group 5 |
| Systemic disorders (including sarcoidosis, pulmonary Langerhans cell histiocytosis, and neurofibromatosis type 1) | |
| Metabolic disorders (including glycogen storage diseases and Gaucher disease) | |
| Chronic renal failure | |
| Malignancy | |
| Fibrosing mediastinitis |
PE: pulmonary embolism; PH: pulmonary hypertension.
Laboratory testing is mandatory18 and should assess concomitant organ damage and comorbidities (blood counts, electrolytes, kidney and liver function markers, iron status, thyroid function, and N-terminal pro-brain-type natriuretic peptide [NT-proBNP]). Directed etiologic studies such as HIV and hepatitis serology will enable identification of possible subgroups, and an autoimmune panel should be obtained.18 In patients originating from or having traveled to schistosomiasis endemic areas, microscopic urine and stool examination should be requested and/or serologic tests performed.
An abdominal ultrasound with portal vein Doppler interrogation is recommended, particularly if liver disease is suspected in patients with newly diagnosed PH, to detect liver disease, portal hypertension or portocaval shunts.18
Ventilation–perfusion lung scanning is key to rule out a diagnosis of CTEPH, the main differential diagnosis in precapillary PH, with superior results compared to CTPA performed outside PH expert centers.18
Genetic counseling is taking on an increasingly important role in managing PAH patients, as mutations in PAH genes have been identified in familial PAH, idiopathic PAH (IPAH), anorexigen-associated PAH and PVOD. Patients should be informed about the possibility of a hereditary component, and counseling should be provided by trained professionals.3
Risk assessmentRisk stratification is essential in managing PAH patients, enabling assessment of disease severity, determination of an appropriate treatment strategy, and guidance of therapeutic adjustments. Risk assessment uses multiparametric scores gathering information from symptoms, exercise capacity and RV function.33
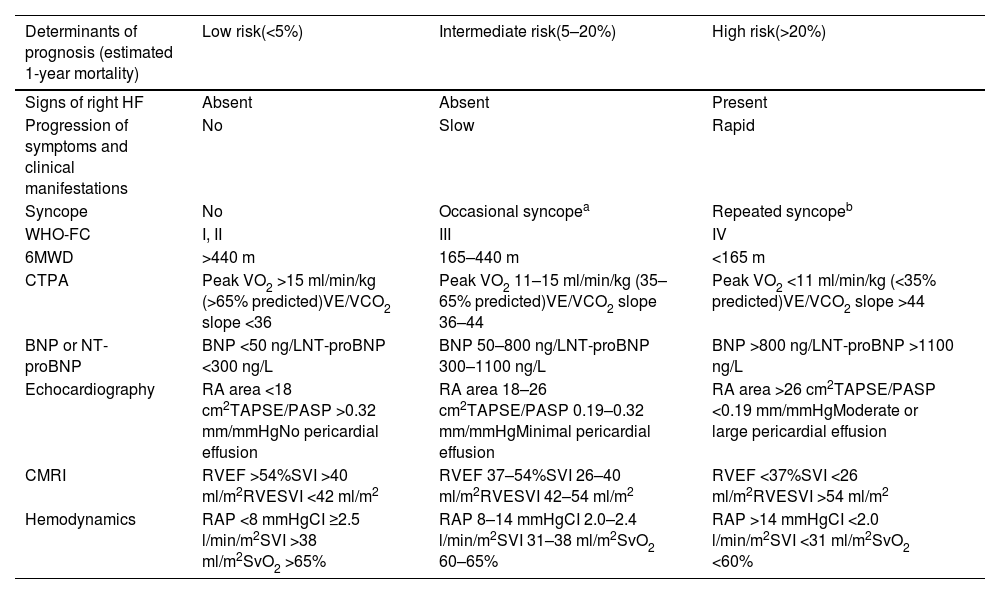
Several scores are currently available, notably COMPERA 2.0,34 the ESC/ERS risk assessment tool28 (Table 6), and the REVEAL 2.0 risk score.35 Different risk scores may have unique strengths and limitations, and the choice of risk assessment tool may vary based on clinician preference and institutional practice.
Comprehensive risk assessment in pulmonary arterial hypertension – three-strata model.
| Determinants of prognosis (estimated 1-year mortality) | Low risk(<5%) | Intermediate risk(5–20%) | High risk(>20%) |
|---|---|---|---|
| Signs of right HF | Absent | Absent | Present |
| Progression of symptoms and clinical manifestations | No | Slow | Rapid |
| Syncope | No | Occasional syncopea | Repeated syncopeb |
| WHO-FC | I, II | III | IV |
| 6MWD | >440 m | 165–440 m | <165 m |
| CTPA | Peak VO2 >15 ml/min/kg (>65% predicted)VE/VCO2 slope <36 | Peak VO2 11–15 ml/min/kg (35–65% predicted)VE/VCO2 slope 36–44 | Peak VO2 <11 ml/min/kg (<35% predicted)VE/VCO2 slope >44 |
| BNP or NT-proBNP | BNP <50 ng/LNT-proBNP <300 ng/L | BNP 50–800 ng/LNT-proBNP 300–1100 ng/L | BNP >800 ng/LNT-proBNP >1100 ng/L |
| Echocardiography | RA area <18 cm2TAPSE/PASP >0.32 mm/mmHgNo pericardial effusion | RA area 18–26 cm2TAPSE/PASP 0.19–0.32 mm/mmHgMinimal pericardial effusion | RA area >26 cm2TAPSE/PASP <0.19 mm/mmHgModerate or large pericardial effusion |
| CMRI | RVEF >54%SVI >40 ml/m2RVESVI <42 ml/m2 | RVEF 37–54%SVI 26–40 ml/m2RVESVI 42–54 ml/m2 | RVEF <37%SVI <26 ml/m2RVESVI >54 ml/m2 |
| Hemodynamics | RAP <8 mmHgCI ≥2.5 l/min/m2SVI >38 ml/m2SvO2 >65% | RAP 8–14 mmHgCI 2.0–2.4 l/min/m2SVI 31–38 ml/m2SvO2 60–65% | RAP >14 mmHgCI <2.0 l/min/m2SVI <31 ml/m2SvO2 <60% |
6MWD: 6-min walk distance; BNP: brain-type natriuretic peptide; CI: cardiac index; CMRI: cardiac magnetic resonance imaging; CTPA: computed tomography pulmonary angiography; NT-proBNP: N-terminal pro-brain-type natriuretic peptide; PASP: pulmonary artery systolic pressure; RA: right atrial; RAP: right atrial pressure; RVEF: right ventricular ejection fraction; RVESVI: right ventricular end-systolic volume index; SVI: stroke volume index; SvO2: mixed venous oxygen saturation; TAPSE: tricuspid annular plane systolic excursion; VE/VCO2: ventilatory equivalent for carbon dioxide; VO2: oxygen uptake; WHO-FC: World Health Organization functional class.
In terms of clinical assessment, signs of right heart failure, rapid progression of symptoms and repeated syncope are markers of higher risk.
Regarding functional status, the World Health Organization functional class (WHO-FC) scale is a simple yet valuable tool for both diagnosis and follow-up, and is one of the strongest indicators of disease progression.36,37
The 6-min walk distance (6MWD) test is the most widely used measure of exercise capacity, due to reproducibility, ease of use and widespread validation. Change in 6MWD is one of the most commonly used parameters in PAH clinical trials as a clinical endpoint.
Cardiopulmonary exercise testing (CPET) provides a more detailed assessment of exercise capacity, peak oxygen consumption, and ventilatory efficiency. The prognostic value of peak VO2 and VE/VCO2 has been established in several studies.38–40
BiomarkersThere have been attempts to identify novel biomarkers to guide prognosis, help with differential diagnosis and assess treatment response. However, brain-type natriuretic peptide (BNP) and NT-proBNP remain the only routinely used biomarkers. They are a nonspecific measure of myocardial stress and cavity dilation, providing prognostic information.41
ImagingImaging delivers reliable assessment of RV function, through direct and indirect measurements.
Echocardiography is the reference imaging method. RV dysfunction can be assessed by measuring various parameters: fractional area change, TAPSE, tissue Doppler, and two-dimensional speckle-tracking myocardial strain recording of RV free wall motion. Maladaptation and RV overload are indirectly revealed by measurements such as inferior vena cava dilation, pericardial effusion, and worsening tricuspid regurgitation.42–45 Recently, combined parameters have been used, such as the TAPSE/PASP ratio, which is a measure of RV-PA coupling and can predict outcomes.46
Echocardiography enables measurements of RV and right atrial (RA) size, which combined with the left ventricular (LV) eccentricity index are valuable in assessing reverse RV remodeling, and can be used as a marker of treatment efficacy.47
Cardiac magnetic resonance imaging (CMRI) provides safe and reproducible cardiac assessment, with the most accurate characterization of RV function and anatomy. The most common abnormalities seen in PH include reduced RV ejection fraction, RV hypertrophy (due to chronic pressure overload), RA and RV enlargement, flattening or leftward bowing of the interventricular septum, dilated pulmonary arteries, tricuspid regurgitation, and pericardial effusion. In addition, CMRI can be used to quantity blood flow in the PA, aorta, and vena cava, enabling quantification of stroke volume and assessment of intracardiac shunts. CMRI thresholds can be used to risk stratify PAH patients according to the ESC/ERS ‘traffic light’ system at baseline and follow-up. RV ejection fraction, stroke volume index and RV end-systolic volume index are three variables that have demonstrated an independent correlation with mortality.
Right heart catheterizationPVR measurement is essential to the diagnosis and prognosis of PH. PVR ≥3.0 WU is associated with poor prognosis.32
Elevated right atrial pressure (RAP) reflects RV pressure overload and is an established risk factor for mortality in PH. A mean RAP >20 mmHg is a proven independent predictor for worse prognosis.48
Acute pulmonary vasoreactivity tests are useful in identifying potential responders to high-dose calcium channel blockers (CCBs), and are conducted in idiopathic, drug-induced, or hereditary PAH patients. PAH patients with other etiologies have a low probability of responding to this therapy.49
mPAP provides little prognostic information, except in acute vasodilator responders.50 Stroke volume index (SVI) has been added to the latest risk stratification table of the ESC/ERS guidelines,18 to determine low- and high-risk status.
Hemodynamic parameters are an important component of risk scores: RAP and PVR in the REVEAL risk scores35 and RAP, cardiac index (CI), and SvO2 in the ESC/ERS risk stratification table.18
The optimal timing of follow-up RHC has not been determined, and varies between centers and patients. However, its importance in the management of PAH cannot be overstated. RHC is a highly informative exam in terms of prognosis and response to therapy. Nonetheless, it carries the potential risks of an invasive exam. Robust data have demonstrated that noninvasive parameters enable adequate prognostic stratification in most PAH cases, often obviating the need for repeat RHC.
Risk stratification in PAH patients presents inherent challenges and pitfalls. Despite its crucial role in guiding treatment decisions, the process relies on multiple parameters, each with its own limitations. Factors such as the dynamic nature of PAH, individual patient responses, and the influence of comorbidities introduce complexity and potential inaccuracies. Moreover, the evolving landscape of PAH therapies and the need for multidimensional assessment introduce even more complexity into the process. Misinterpreting or overlooking certain variables may lead to suboptimal risk assessments, impacting treatment strategies and patient outcomes. The ongoing efforts to refine risk stratification models underscore the need for a nuanced and adaptable approach that considers the intricacies of individual cases in the complex realm of PAH.
Screening and early detectionDue to the development of increasingly effective therapeutic options, early diagnosis proves key in improving patient outcomes.
Groups known to have high-risk traits, such as individuals with SSc, BMPR2 mutation carriers, first-degree relatives of patients with hereditary PAH, and patients being assessed for liver transplantation, benefit from screening for PAH.18 Screening tools such as blood biomarkers, electrocardiography, echocardiography, pulmonary function tests, and exercise testing are valuable for assessing these patients,51 even though the optimal interval for each subgroup has not been established.
Practical approach to risk stratificationCurrent guidelines support a risk-based, goal-orientated approach, in which achieving and maintaining a low risk status is recommended.18
At diagnosis, the three-strata model (Table 6) is recommended, considering as many factors as possible, to classify patients according to one-year mortality risk as low (<5%), intermediate (5–20%), or high (>20%) risk.
For follow-up, a four-strata model (Table 7) has been developed, enabling a finer stratification into four groups, based on simpler clinical and laboratory assessments. It subdivides the intermediate group, in which most patients are classified, into two subgroups. It should be reassessed at each subsequent visit, every three to six months.
Simplified four-strata risk assessment tool.
| Determinants of prognosis | Low risk | Intermediate–low risk | Intermediate–high risk | High risk |
|---|---|---|---|---|
| Points assigned | 1 | 2 | 3 | 4 |
| WHO-FC | I or IIa | – | III | IV |
| 6MWD, m | >440 | 320–440 | 165–319 | <165 |
| BNPor NT-proBNP, ng/L | <50<300 | 50–199300–649 | 200–800650–1100 | >800>1100 |
6MWD: 6-min walk distance; BNP: brain-type natriuretic peptide; NT-proBNP: N-terminal pro-brain-type natriuretic peptide; WHO-FC: World Health Organization functional class.
Risk is calculated by dividing the sum of all grades by the number of variables and rounding to the next integer.
Currently approved therapies can be divided into three major categories according to their therapeutic target: prostacyclins (cyclic adenosine monophosphate [cAMP] pathway), phosphodiesterase (PDE)-5 inhibitors and soluble guanylate cyclase stimulators (nitric oxide [NO]-cyclic guanosine monophosphate [cGMP] pathway), and endothelin receptor antagonists (ERAs) (endothelin-1 [ET-1] pathway). In addition, CCBs can be utilized in a specific subset of patients (Figure 1).
These therapies rely on the premise of vasodilation and have been the mainstay approach in recent decades. Their use has significantly improved quality of life, functional status, and survival.18 They do not, however, address the fundamental processes that initiate and perpetuate the disease, and have little effect on fibrotic and proliferative changes, and so have no curative potential.
Calcium channel blockersCCBs work by blocking the influx of calcium ions into smooth muscle cells lining the arterial wall, leading to vasodilation. This therapy is reserved for a select group of patients who respond favorably to acute vasodilator challenge during RHC. The criterion for a positive response to acute vasodilator challenge – with inhaled NO (preferably), iloprost or intravenous (IV) epoprostenol1 – is a reduction in mPAP of ≥10 mmHg to reach an absolute value of ≤40 mmHg, with increased or unchanged cardiac output. Most PH patients present a minimal response. Some, however, demonstrate vasodilation to normal or near normal hemodynamics. These are identified as acute responders and have a better prognosis after initiation of appropriate treatment with high doses of CCBs. Long-term response is significantly better for idiopathic, hereditary and drug/toxin-associated PAH patients, the only groups in which the vasodilation test should be prescribed at the time of RHC.50,52
Dihydropyridines (DHPs), such as amlodipine, nifedipine and felodipine, have exclusively vascular effects, reducing unwanted cardiodepressant activity. The most common side effects are related to their vasodilatory properties: headache, flushing, dizziness, nausea, and lower extremity edema.53 Non-DHPs such as diltiazem are preferred in patients with a resting HR of >80 bpm, due to their cardiodepressant effect, but are contraindicated in cases of systolic dysfunction and hemodynamic instability.
Nitric oxide pathwayNO is produced in PAECs by endothelial nitric oxide synthase. It diffuses into the underlying PASMCs, binding to soluble guanylate cyclase (sGC), converting guanosine triphosphate to cGMP. The subsequent activation of cGMP-dependent protein kinases results in vasodilation. Additionally, NO inhibits PASMC proliferation, platelet aggregation, and thrombosis.10
Phosphodiesterase 5 inhibitors (PDE-5is), such as sildenafil and tadalafil, prevent the degradation of cGMP, promoting the above-mentioned antiproliferative and vasodilatory effects of NO.
Adverse effects of these drugs include headaches, epistaxis, flushing, dyspepsia, and diarrhea, with varying severity. They should be used with care in combination with other vasodilators, such as nitrates and CCBs, to avoid hypotension.
Sildenafil has been shown to significantly improve results in the 6MWD test, WHO-FC and PAP compared to placebo, with no significant difference in clinical worsening, as shown in the SUPER-1 and SUPER-2 trials.54,55 It is predominantly used in its oral formulation but can be administered intravenously for long-term patients who are unable to tolerate oral forms.
Tadalafil is an alternative that has the advantage of a single daily 40-mg administration, compared to sildenafil 20 mg three times daily. In the PHIRST-1 trial,56 it showed significant improvement in the 6MWD test compared to placebo in treatment-naïve patients.
Guanylate cyclase stimulators directly activate sGC, promoting vasodilation. In the PATENT-1 trial57 and the open-label extension PATENT-2,58 riociguat was shown to significantly improve exercise capacity and WHO-FC, as well as reducing PVR, NT-proBNP levels and time to clinical worsening. The REPLACE trial,59 which investigated the efficacy and safety of switching from a PDE-5i to riociguat compared with remaining on the PDE-5i in patients with PAH at intermediate risk, favored riociguat regarding clinical improvement. The side-effect profile of riociguat is similar to that of PDE-5is.
Prostacyclin-thromboxane A2 pathwayProstacyclins, produced in PAECs from arachidonic acid, activate adenylate cyclase in smooth muscle cells by binding to prostanoid receptors, leading to smooth muscle relaxation. They also inhibit platelet aggregation and smooth muscle proliferation, and have anti-inflammatory and antithrombotic effects. TXA2, on the other hand, promotes vasoconstriction and platelet aggregation.60
Prostacyclin analogsEpoprostenol has a short half-life and limited stability at room temperature, and can only be administered in continuous intravenous infusions. Randomized trials have shown the benefits of this drug in symptomatic control, exercise capacity and hemodynamics.61,62 It has shown improved survival rates in patients with severe IPAH.62
Treprostinil has similar properties and benefits,63 and its longer half-life means different routes of administration can be used. The subcutaneous route improves exercise capacity, hemodynamics, and symptoms in PAH63 but infusion site pain is a common adverse effect, leading to patient dissatisfaction. This challenge can be addressed through IV administration, particularly when facilitated by an implanted pump system, ensuring sustained and consistent drug delivery. Inhaled treprostinil was investigated in the TRIUMPH trial,64 with significant improvement in 6MWD and quality of life in patients already receiving bosentan or sildenafil. Oral formulations have been studied in the FREEDOM-C65 and FREEDOM-C266 trials, also in addition to bosentan and/or sildenafil therapy. Both studies failed to demonstrate improvements in 6MWD. A subsequent randomized clinical trial with treatment-naïve patients showed a significant improvement.67 The oral route of administration is not approved in Europe.
Iloprost is available in IV or aerosol formulations. Its inhaled route of administration was demonstrated to confer significant improvements in symptomatic control, hemodynamics, WHO-FC and quality of life in the AIR study.68 The most common side effects noted were flushing and jaw pain. Data concerning IV and oral iloprost in PAH are scarce; an observational study demonstrated limited clinical benefits.69
Prostacyclin receptor agonistsSelexipag is an oral prostacyclin receptor IP agonist. Its efficacy was studied in the GRIPHON study, which involved PAH patients with or without pre-existing mono or dual therapy.70 There was a reduction of about 40% in time to first morbidity or mortality event, though mortality rates were not significantly affected. 6MWD was significantly improved. The most common adverse effects were headache, diarrhea, nausea, and jaw pain.70
Ralinepag is another oral prostacyclin receptor IP agonist. Its efficacy and safety are being evaluated in the ongoing ROR-PH-301 trial (clinicaltrials.gov identifier: NCT03626688), when added to PAH standard of care or PAH-specific background therapy.
Endothelin pathwayThe endothelin system, comprising endothelin-1 (ET-1) and its receptors (ET-A and ET-B), plays a pivotal role in pulmonary vascular homeostasis. ET-A receptors are most commonly found on vascular smooth muscle cells and induce vasoconstriction by increasing intracellular calcium. ET-B receptors are predominantly located on endothelial cells, which stimulate the release of vasodilating agents such as NO and prostacyclin.71 ET-B receptors also appear on vascular smooth muscle cells, stimulating vasoconstriction. Theoretically, either selective blocking of ET-A receptors alone, or non-selective blocking of both ET-A and ET-B receptors together, could dilate local pulmonary vessels.
Endothelin receptor antagonistsERAs act as competitive antagonists at the endothelin receptors and have shown promise in countering the deleterious effects of ET-1.
Bosentan is a dual-acting ERA, causing non-selective antagonism of both ET-A and ET-B receptors. Its use has led to significant improvements in 6MWD, functional class, and time to first worsening, with results published from BREATHE-1,72 a double-blind, placebo-controlled trial in which patients were randomly assigned to receive 62.5 mg of bosentan twice daily for four weeks followed by either of two doses of bosentan (125 or 250 mg twice daily) for a minimum of 12 weeks. These findings were similarly demonstrated in a subsequent 24-week study, with both trials showing increased liver enzymes as the main adverse event.73 Dose-dependent increases in liver transaminases can occur in up to 10% of treated patients and are reversible after dose reduction or discontinuation.74 Monthly liver function testing is advised for patients receiving bosentan, which may also render hormonal contraceptives unreliable and lower serum levels of warfarin, sildenafil and tadalafil.
Ambrisentan is an oral ERA that binds selectively to ET-A receptors. In the ARIES randomized double-blind controlled trials,75 treatment-naïve PAH patients who were started on ambrisentan showed improvements in 6MWD, functional class and time to clinical worsening. The recorded adverse effects included peripheral edema, headache, and nasal congestion.
Macitentan is a dual ERA, evaluated in the pivotal SERAPHIN trial,76 a randomized, double-blind, placebo-controlled study. Macitentan-treated patients exhibited a 45% reduction in the risk of reaching the primary endpoint (a composite of PAH-related death, hospitalization for PAH, and disease progression). It had a favorable safety profile, with no significant differences in adverse events compared to placebo. The most frequently reported adverse events were headache, anemia, and nasopharyngitis. Its clinical efficacy and tolerability have been supported by real-world studies and post-marketing surveillance.
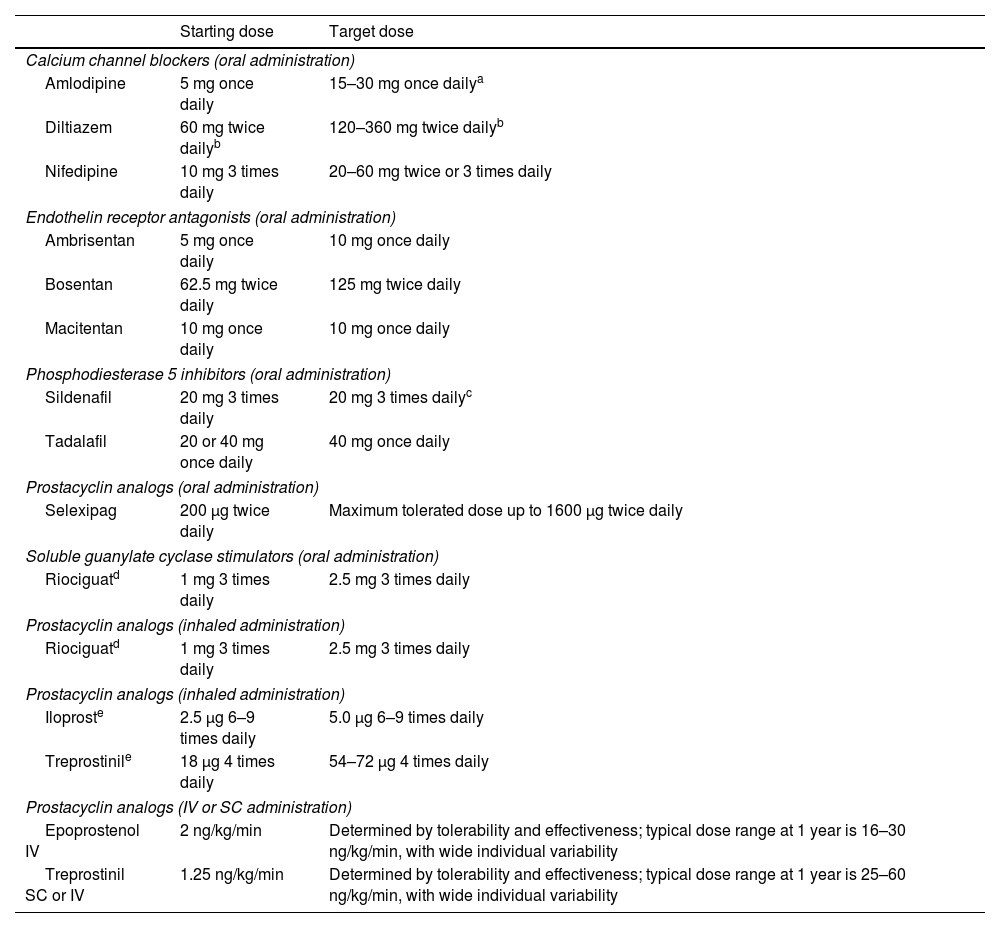
Therapeutic approachTreatment strategies have evolved in parallel with advances in the development of novel agents. Current guidelines18 recommend adapting the initial strategy according to disease etiology, comorbidities, and risk assessment. In addition to targeted drug therapy, which will be discussed below, general measures can be applied to all patients, regardless of subtype (Table 8 and Figure 7).
Dosing of pulmonary arterial hypertension medication in adults.
| Starting dose | Target dose | |
|---|---|---|
| Calcium channel blockers (oral administration) | ||
| Amlodipine | 5 mg once daily | 15–30 mg once dailya |
| Diltiazem | 60 mg twice dailyb | 120–360 mg twice dailyb |
| Nifedipine | 10 mg 3 times daily | 20–60 mg twice or 3 times daily |
| Endothelin receptor antagonists (oral administration) | ||
| Ambrisentan | 5 mg once daily | 10 mg once daily |
| Bosentan | 62.5 mg twice daily | 125 mg twice daily |
| Macitentan | 10 mg once daily | 10 mg once daily |
| Phosphodiesterase 5 inhibitors (oral administration) | ||
| Sildenafil | 20 mg 3 times daily | 20 mg 3 times dailyc |
| Tadalafil | 20 or 40 mg once daily | 40 mg once daily |
| Prostacyclin analogs (oral administration) | ||
| Selexipag | 200 μg twice daily | Maximum tolerated dose up to 1600 μg twice daily |
| Soluble guanylate cyclase stimulators (oral administration) | ||
| Riociguatd | 1 mg 3 times daily | 2.5 mg 3 times daily |
| Prostacyclin analogs (inhaled administration) | ||
| Riociguatd | 1 mg 3 times daily | 2.5 mg 3 times daily |
| Prostacyclin analogs (inhaled administration) | ||
| Iloproste | 2.5 μg 6–9 times daily | 5.0 μg 6–9 times daily |
| Treprostinile | 18 μg 4 times daily | 54–72 μg 4 times daily |
| Prostacyclin analogs (IV or SC administration) | ||
| Epoprostenol IV | 2 ng/kg/min | Determined by tolerability and effectiveness; typical dose range at 1 year is 16–30 ng/kg/min, with wide individual variability |
| Treprostinil SC or IV | 1.25 ng/kg/min | Determined by tolerability and effectiveness; typical dose range at 1 year is 25–60 ng/kg/min, with wide individual variability |
IV: intravenous; SC: subcutaneous.
The daily dosages of amlodipine and felodipine can be administered in a single dose or divided into two doses.
There are different release formulations of diltiazem, some of which should be administered once or 3 times daily.

Pulmonary arterial hypertension treatment algorithm.
a Cardiopulmonary comorbidities are conditions associated with an increased risk of left ventricular diastolic dysfunction, and include obesity, hypertension, diabetes, and coronary heart disease; pulmonary comorbidities may include signs of mild parenchymal lung disease and are often associated with a low DLCO.
b Intravenous epoprostenol or intravenous/subcutaneous treprostinil.
DLCO: diffusion capacity of the lung for carbon monoxide; ERA: endothelin receptor antagonist; PCA: prostacyclin analog; PDE-5i: phosphodiesterase 5 inhibitor; PRA: prostacyclin receptor agonist; sGCs: soluble guanylate cyclase stimulator.
Adapted from the 2022 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension.1
Preventing situations that could lead to right heart decompensation is paramount. It is recommended that patients with PAH receive vaccinations against Streptococcus pneumoniae, influenza, and the SARS-CoV-2 virus.18
Optimization of volume status with diuretics is important in patients with RV dysfunction to reduce symptoms of overload and improve hemodynamics.77 Particular attention is recommended to correction of anemia and iron deficiency. Supplementary oxygen should be provided to patients with respiratory insufficiency. Exercise training programs should be encouraged in patients who can tolerate them and adapted to patients with worse functional capacity. Psychosocial support should be made available, and patients should be provided with information regarding prognosis, if they so desire. Choices regarding treatment decisions should be shared with properly informed patients, as with all decisions in modern practice.
For vasodilator responders, initial therapy with high-dose CCBs should be initiated. These patients are to be closely monitored, with a complete reassessment after 3–6 months of therapy, including RHC. Additional acute vasoreactivity testing should be performed at reassessment to confirm sustained vasodilator response.1 Patients deemed to have a satisfactory chronic response are those who present with WHO-FC I/II and hemodynamic improvement (ideally, mPAP <30 mmHg and PVR <4 WU). In the absence of a satisfactory response, additional PAH therapy should be instituted (Figure 7).
Most clinical trials study populations with idiopathic, heritable, drug or connective tissue disease-associated PAH. This section will discuss therapeutic management of these patients (Figure 7).
Combination therapies have been progressively adopted as an early option in the treatment of PAH as they enable multiple signaling pathways to be targeted. Meta-analyses have shown reductions in time to clinical worsening and improvements in clinical outcomes when sequential combination therapy was added following suboptimal response to monotherapy,78 and benefits of combination therapy over monotherapy.79
Regarding initial strategy, AMBITION80 was a pivotal trial in combination therapy, comparing initial dual therapy with ambrisentan and tadalafil versus monotherapy with either ambrisentan or tadalafil, in treatment-naïve patients. In a follow-up of 73 weeks, a 50% risk reduction was recorded with initial dual therapy, for the primary endpoint of time to clinical event (composite of death, hospitalization for worsening PAH, disease progression, or unsatisfactory long-term clinical response). There were also significant improvements in 6MWD and NT-proBNP with initial combination therapy.
Bearing in mind the pathophysiology of PAH, initial triple therapy would logically result in even better outcomes. However, this was not the case in the TRITON study,81 which compared initial triple-combination therapy with macitentan, tadalafil and selexipag to initial dual therapy with macitentan and tadalafil in treatment-naïve patients. There were no significant differences between the groups regarding PVR (reductions of 54% and 52% respectively), 6MWD (increases of 56 m and 55 m) and mean NT-proBNP. This trial did not show benefit in initial triple therapy but did reinforce evidence supporting initial ERA/PDE-5i combination therapy.
Data from the French registry,82 however, showed that initial triple-combination therapy including an IV/SC prostacyclin analog was associated with better long-term survival than monotherapy or dual combination therapy. More studies are required to clarify whether initial triple therapy is a superior approach, and in which patients it should be initiated.
The current guidelines18 stratify patients according to risk (see section “Risk assessment”) and the presence or absence of cardiovascular comorbidities associated with an increased risk of LV diastolic dysfunction (such as obesity, hypertension, diabetes, or ischemic heart disease) or pulmonary comorbidities, to guide therapy. In a population as complex as PH patients, however, treatment decisions must be personalized, going beyond simplified algorithms.
Patients without significant comorbiditiesFor patients with low or intermediate risk, initial combination therapy with an ERA and a PDE-5i is recommended. In patients presenting at high risk, initial triple-combination therapy including an IV/SC prostacyclin analog should be considered (Table 8). Initial triple-combination therapy should also be considered in patients at intermediate risk presenting with severe hemodynamic impairment (RAP >14 mmHg, CI <2.0 l/min/m2, SVI <31 ml/m2, and/or PVR ≥12 WU).
Patients with significant comorbiditiesTreating patients with PAH who also present with comorbidities poses a significant challenge, sparking debates and controversies within the medical community. Concurrent conditions such as left heart failure, lung disease or SSc makes the optimal treatment strategy contentious. Balancing the need for aggressive PAH-specific therapies against potential complications and interactions with medications used for comorbidities requires careful consideration. These controversies underscore the critical need for further research, consensus-building, and personalized treatment plans to navigate the complexities of managing PAH in patients with diverse medical backgrounds.
For patients with significant cardiovascular or pulmonary comorbidities, oral monotherapy (PDE-5i or ERA) should be preferred regardless of mortality risk (Figure 7). Although evidence supporting the superiority of a particular molecule may be lacking, most patients in this group are treated with PDE-5i monotherapy.83
Approach to advanced diseaseEligible patients who remain at intermediate–high or high risk on optimal therapy should be referred for lung transplantation.18
Other interventional options may be considered to decrease RV afterload. These include atrial septostomy or a Potts shunt (an anastomosis between the left pulmonary artery and the descending aorta), which can improve hemodynamics and symptoms of right heart failure as a palliative procedure or as a bridge to transplantation.84,85 Venoarterial extracorporeal membrane oxygenation is being increasingly used as rescue therapy or temporary support.
Finally, all patients with PAH who are eligible and willing to participate should be considered for novel clinical trials.
Novel therapiesAs research advances, the focus has been on acting upstream in the pathophysiological chain, with the objective of not only halting disease progression earlier, but also attempting to reverse tissue damage and remodeling.
Genetic pathways are known to play a critical role in the pathogenesis of PAH. Mutations in BMPR2 have been identified as the most frequent cause of heritable PAH.4 This pathway is also involved in the development of PAH in patients without heritable disease.86
Sotatercept is a first-in-class fusion protein that acts as a ligand trap for members of the TGF-β superfamily, thus restoring balance between the growth-promoting activin growth differentiation factor pathway and the growth-inhibiting bone morphogenetic protein (BMP) pathway. In PULSAR, a randomized, double-blind, placebo-controlled phase 2 trial, subcutaneous administration of sotatercept met the primary endpoint assessed at week 24, with a significant reduction of 34% in PVR compared to placebo, in addition to approved background therapy.87 Efficacy for secondary endpoints including 6MWD, NT-proBNP, RAP and mPAP was also shown. The phase 3 multicenter double-blind STELLAR trial88 that included 323 patients with PAH on background therapy showed a significant change in the primary endpoint (exercise capacity as assessed by 6MWD) and eight out of nine secondary endpoints. The main adverse events were thrombocytopenia, increase in hemoglobin and telangiectasia (in about 10% of patients).
KER-012 is a novel molecule, designed to bind to and inhibit the signaling of TGF-β ligands, including activin A, activin B and myostatin, potentially increasing the signaling of BMP pathways. TROPOS, a randomized, double-blind, placebo-controlled phase 2 clinical trial, is currently underway to evaluate KER-012 in combination with background therapy in patients with PAH. Outcomes will include changes in PVR, 6MWD, and safety parameters.
The platelet-derived growth factor-mediated pathway, which promotes the proliferation of PASMCs and an inflammatory environment, has also been identified as a therapeutic target.89
Imatinib, a tyrosine kinase inhibitor currently used to treat multiple cancers, was the first antiproliferative to have demonstrated clinical efficacy in phase 2 and 3 trials of PAH when administered orally. However, systemic side effects limited its therapeutic use.90 The IMPAHCT study, a currently ongoing phase 2B/3 trial, will evaluate the safety, tolerability, and clinical benefit of inhaled imatinib in PAH.
Seralutinib, another tyrosine kinase inhibitor, acts through the same mechanism, and has been evaluated in the TORREY phase 2 study, in which it led to a reduction of 14% in PVR and significant reductions in NT-proBNP levels and improvement in 6MWD and functional class.91
There are a myriad other therapeutic targets and ongoing clinical studies, with targets ranging from hormonal pathways (e.g. anastrozole and tamoxifen) to metabolism (bardoxolone, metformin and ranolazine), epigenetic alterations and DNA damage (apabetalone and histone deacetylase inhibitors), the serotonin pathway (serotonin reuptake inhibitors), and vasoactive agents (apelin receptor agonists).
ConclusionPAH is a complex disease, a consequence of vascular dysfunction and remodeling, that stems from multiple possible etiologies. Patients should be managed at high-volume and experienced centers. Nonetheless, diagnosis results from clinical suspicion, and physician awareness is key.
The field of PAH management is ever-changing, and current research brings the promise of increasingly effective and safe options for this high-risk group of patients with a historically poor prognosis.
Conflicts of interestThe authors have no conflicts of interest to declare.
