






A síndroma de QT longo congénita (SQTL) pode manifestar-se por síncopes ou convulsões recorrentes, no contexto de taquicardia ventricular polimórfica, podendo simular epilepsia.
Nos doentes tratados com cardioversor-desfibrilhador implantável (CDI) a recorrência de arritmias com consequente terapêutica com choques frequentes pode conduzir a reacções adversas, nomeadamente psicogénicas.
Apresentamos o caso de uma doente de 22 anos com síncopes e crises convulsivas, cujo diagnóstico era desde a infância de epilepsia, e em quem a SQTL foi diagnosticada apenas em idade adulta. Por falência da terapêutica beta-bloqueante implantou CDI, e por persistência de arritmias foi submetida a simpaticectomia cardíaca esquerda. O follow-up pós-cirurgia aos 3 meses mostrou redução significativa do número de arritmias.
O estudo genético identificou uma mutação patogénica no gene KCNH2 (SQTL tipo 2), em heterozigotia, a mutação c.1817C >T p.S606F, ainda não descrita na literatura. Relatamos também a rara ocorrência de tempestade arrítmica no contexto de infecção a H1N1.
O caso clínico ilustra as dificuldades quer do diagnóstico quer do tratamento da SQTL. É discutida a possibilidade duma base genética partilhada entre a doença disrítmica e neurológica.
Congenital long QT syndrome (LQTS) can present as syncope or seizures, secondary to polymorphic ventricular tachycardia, mimicking a primary seizure disorder.
In patients treated with an implantable cardioverter-defibrillator (ICD), the recurrence of arrhythmias with subsequent frequent therapeutic shocks may cause adverse reactions, which can be psychogenic.
We report the case of a 22-year-old woman with syncope and seizures who was diagnosed in childhood as epileptic and in whom LQTS was diagnosed only in adulthood. Beta-blocker therapy failed and an ICD was implanted. However, as arrhythmias persisted, left cardiac sympathetic denervation was performed. After surgery, three-month follow-up showed a significant reduction in arrhythmias.
The genetic study identified a heterozygous mutation, c.1817 C>T p.S606F, on the KCNH2 gene that has not previously been reported in the literature. We also report the rare occurrence of an electrical storm in the course of H1N1 infection.
This case illustrates the difficulties in the diagnosis and treatment of LQTS. The possibility of a common genetic basis for arrhythmic diseases and epilepsy is discussed.
A síndroma de QT longo congénita (SQTL) é uma doença arritmogénica hereditária caracterizada por um intervalo QT prolongado, com grande vulnerabilidade para arritmias ventriculares graves, taquicardia ventricular polimórfica do tipo torsade de pointes, manifestando-se por síncope e morte súbita (MS)1. A SQTL é uma canalopatia devida a mutações nos genes que codificam proteínas constituintes dos canais iónicos da membrana celular cardíaca1. Estão identificados 12 genes responsáveis, o que confere grande heterogeneidade genotípica e fenotípica, sendo frequentemente sub-diagnosticada2. As crises convulsivas são também uma forma de apresentação, podendo mascarar o diagnóstico com epilepsia e levar à instituição de terapêutica anti-epiléptica3.
A consequência de uma SQTL não diagnosticada pode ser catastrófica. A SQTL quando não tratada tem uma mortalidade significativa, tendo sido identificados como factores de mau prognóstico: a presença de síncopes em idade inferior a 18 anos, sexo feminino, intervalo QT corrigido (QTc) ≥ 500ms e mutação associada à SQTL tipo 24. A prevenção da morte súbita assenta principalmente na instituição de terapêutica beta-bloqueante, podendo passar também pela implantação de um cardioversor-desfibrilhador implantável (CDI)5. Nos doentes com falência de terapêutica beta-bloqueante, a persistência de arritmias, com consequente cardioversão/ desfibrilhação automática repetida, conduz a um stress contínuo, assim como a inaptidão para a vida social6. Neste contexto, a simpaticectomia cardíaca esquerda surge como a derradeira intervenção terapêutica.
A ocorrência de tempestade arrítmica nestes doentes constitui uma complicação grave, com necessidade de rápido reconhecimento e tratamento.
Apresentamos o caso de uma doente com o diagnóstico de epilepsia desde a infância, a quem foi diagnosticada SQTL apenas em idade adulta. A investigação subsequente identificou uma mutação ainda não descrita.
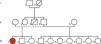
Caso clínicoMulher de 22 anos, de raça negra, com história de síncopes recorrentes desde os dois anos de idade e com o diagnóstico de epilepsia. Apesar de medicada com fenobarbital mantinha síncopes frequentes, por vezes acompanhadas de convulsões tónicas, incontinência de esfíncteres e mordedura de língua. Estes eventos eram precedidos de palpitações rápidas, sendo geralmente desencadeados por emoções e sons intensos. Tinha história familiar de morte súbita, tio paterno (Figura 1).
Pela persistência das síncopes, por vezes acompanhadas de crises convulsivas, foi referenciada a uma consulta de Neurologia para esclarecimento da sua doença. O exame neurológico foi normal, assim como a ressonância magnética crânio-encefálica e o electroencefalograma intercrítico que não mostrou evidência de lentificação focal ou actividade epiléptica.
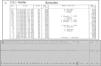
Por não se terem identificado alterações na investigação inicial, o estudo prosseguiu com monitorização por vídeo-electroencefalograma e prova de provocação. Durante o teste verificou-se um episódio sincopal que não se traduziu por actividade epiléptica no registo do electroencefalograma. No entanto, a avaliação do traçado eletrocardiográfico uniderivação mostrou uma taquicardia ventricular polimórfica do tipo torsade de pointes, coincidente com o momento da prova de provocação pela administração de soro fisiológico endovenoso e a ocorrência de síncope (Figura 2).
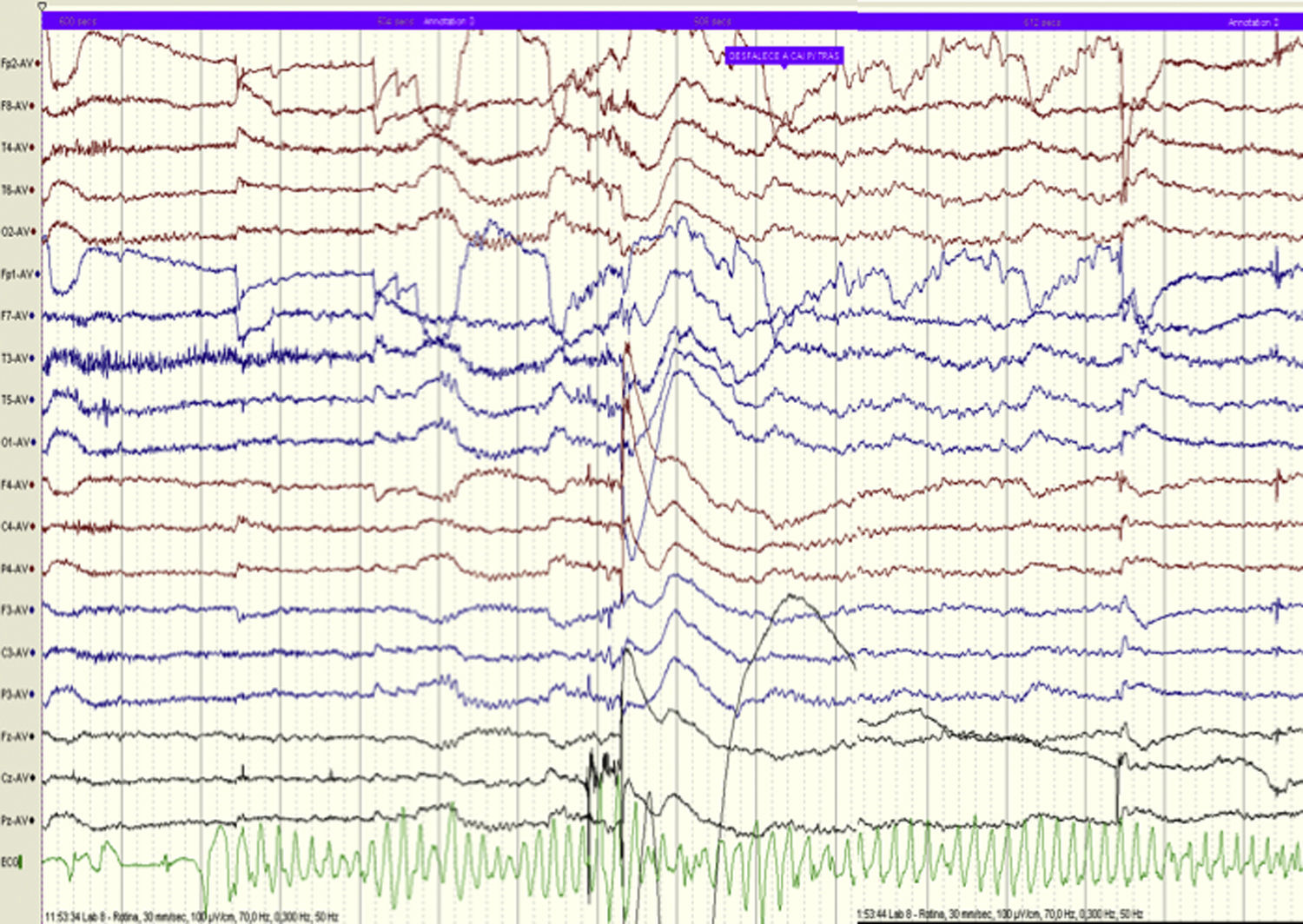
O electrocardiograma de 12 derivações (ECG) mostrava intervalo QTc de 482ms (Figura 3). Realizou ECG de Holter de 24 horas que documentou extrassistolia ventricular frequente, média de 30/h, com 22 pares e 8 salvas de taquicardia ventricular, com dois episódios de torsade de pointes sincopais, de 30 e de 40 segundos. O QTc encontrava-se prolongado: valor médio de 561ms, variando entre 452 e 648ms (Figura 4).
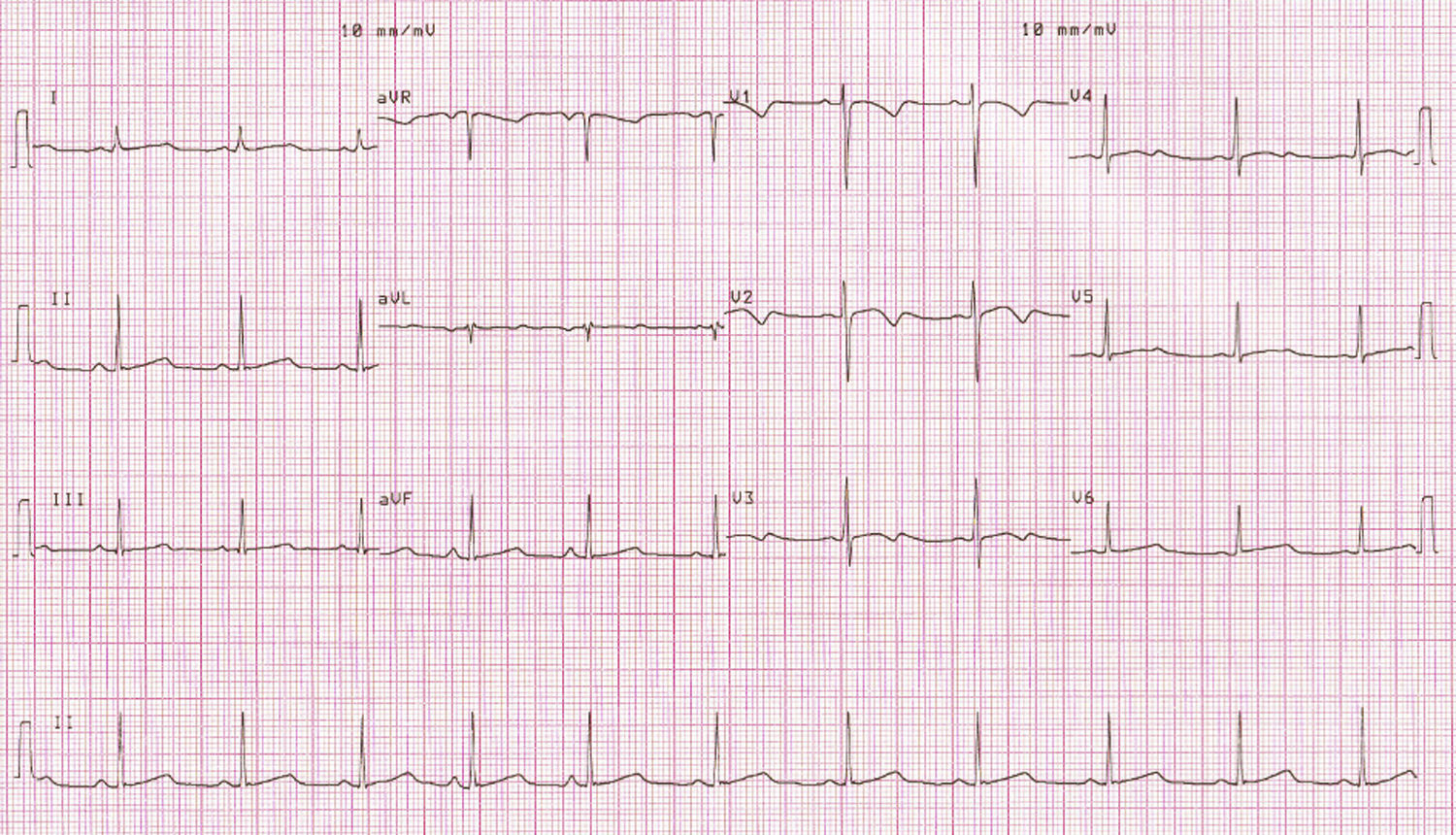
Foi colocado o diagnóstico de síndrome de QT longo, sendo transferida para o Serviço de Cardiologia para monitorização e terapêutica. O exame clínico e o ecocardiograma transtorácico eram normais. Foi colhido sangue para estudo genético.
A atitude terapêutica incluiu a suspensão do fenobarbital, dado o potencial prolongamento do intervalo QT associado a este fármaco e início de propranolol em doses crescentes, até dose máxima tolerável. Apesar destas medidas a doente manteve episódios sincopais precedidas de palpitações rápidas, correspondendo a taquicardia ventricular polimórfica, tendo sido objectivado durante estes episódios a ocorrência de: mídriase, movimentos tónicos, incontinência de esfíncter urinário, respiração ruidosa e sudorese. Nunca houve evidência de sinais neurológicos focais ou confusão pós-ictal. Dada a falência da terapêutica beta–bloqueante nas doses máximas toleráveis decidiu-se, de acordo com as recomendações terapêuticas em vigor, pela implantação de um CDI12. Foi implantado CDI tipo VVI- R modelo Lumax VR® (Biotronic®). Teve alta sob terapêutica beta-bloqueante máxima tolerada.
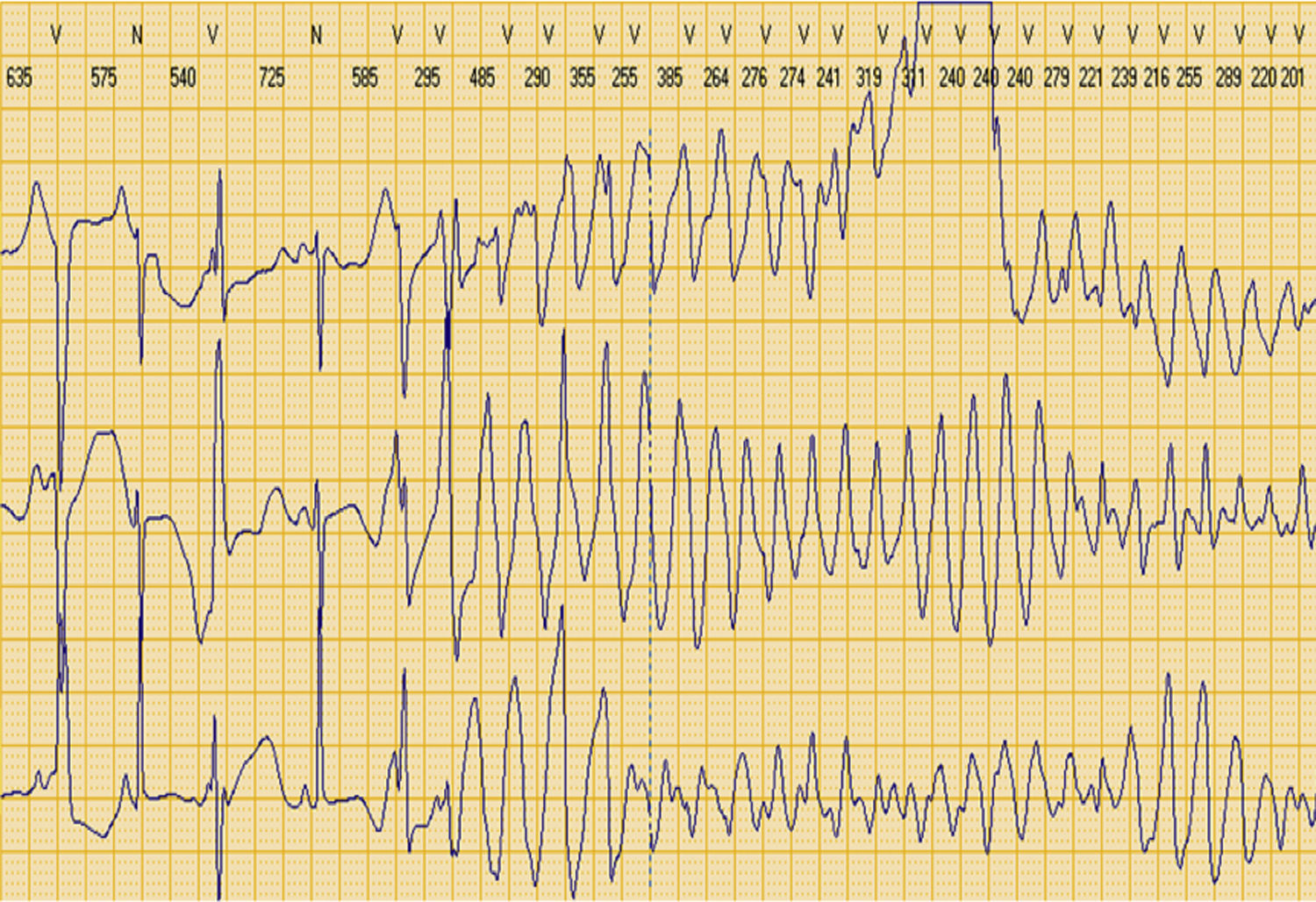
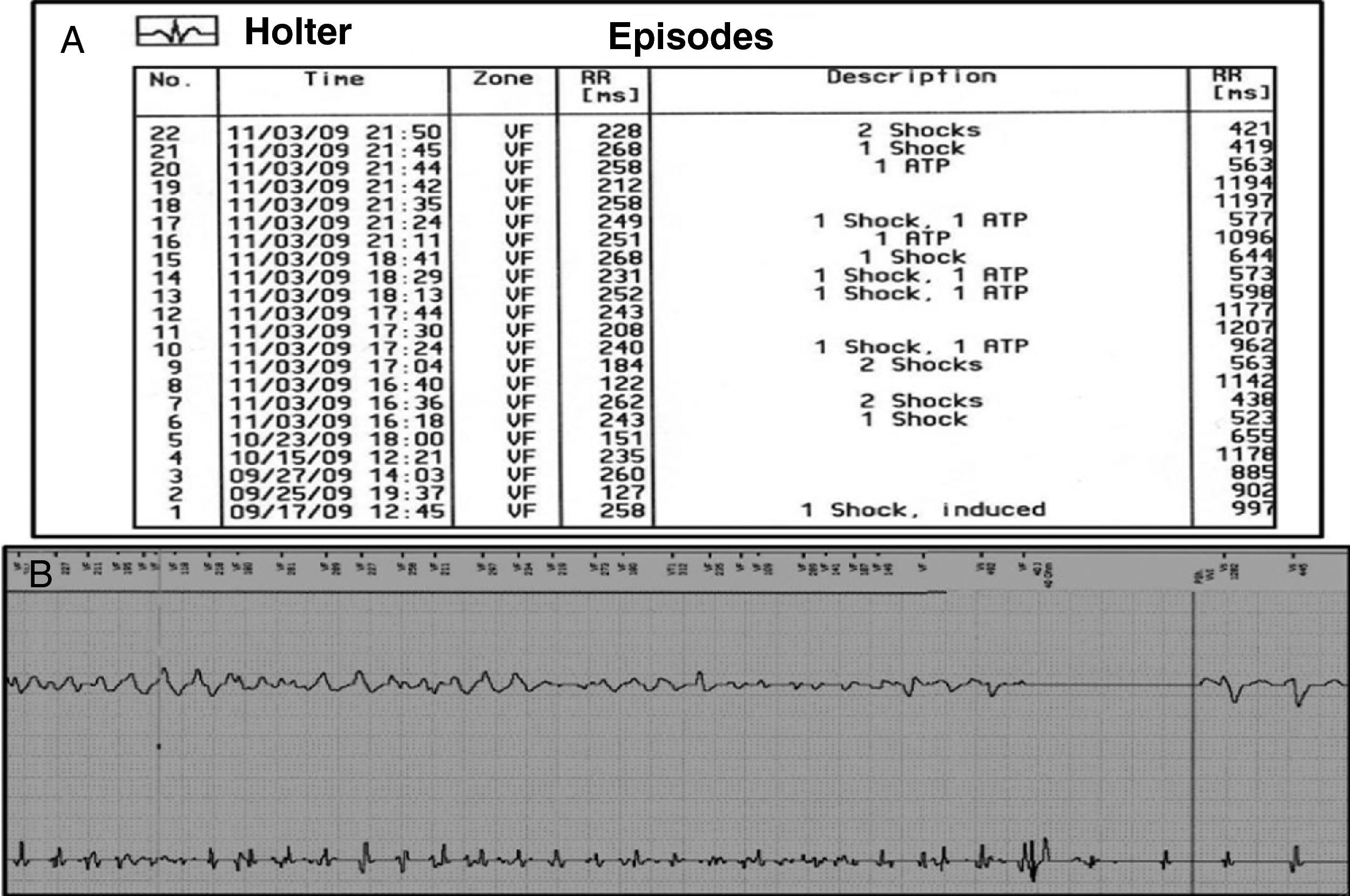
Manteve-se assintomática durante um mês após a alta até que, após dois dias de quadro gripal com febre, teve vários episódios de palpitações rápidas, seguidos de 13 choques apropriados e 6 episódios de pacing anti-taquicardia, como mostrou o registo de eventos do CDI (Figura 6). A investigação da etiologia da tempestade arrítmica excluiu como causas precipitantes: hipomagnesiemia, hipocalcemia, hipocaliemia, suspensão da terapêutica em curso e administração de fármacos que prolongassem o intervalo QT.
Perante o episódio febril e suspeita clínica de infecção gripal foi feita a pesquiza de infecção pelo vírus H1N1, a qual foi confirmada por polymerase chain reaction. Foi excluída miocardite viral e outras possíveis complicações desta infecção. A tempestade arrítmica terminou com o aumento da frequência de pacing ventricular, perfusão de sulfato de magnésio e aumento da dose de propranolol. A evolução favorável do quadro gripal permitiu que não se instituísse terapêutica antiviral com oseltamivir, pelo eventual risco de prolongamento do intervalo QT associado. A doente teve alta mantendo acompanhamento em consulta. O registador de eventos do CDI mostrou várias detecções isoladas de taquicardia ventricular com terapias apropriadas sob terapêutica com propranolol e alprazolam. Para assegurar a adesão terapêutica substituiu-se o propranolol por atenolol na dose de 200mg/dia.
Apesar da terapêutica beta-bloqueante manteve eventos de taquicardia/ fibrilhação ventricular com necessidade de cardioversão/desfibrilhação. Dada a refracteridade à terapêutica foi submetida a simpaticectomia cardíaca esquerda por toracoscopia, que consistiu na remoção do 1/3 inferior do gânglio estrelado e ablação simpática de T2 a T5 com ressecção de ramos colaterais. Houve anidrose do membro superior esquerdo, mas sem Síndrome Horner. No primeiro dia de pós-operatório foi reduzida a dose de atenolol para 100mg/dia, ficando em ritmo próprio. Ao quarto dia de pós-operatório teve episódio de torsade de pointes sincopal. Teve alta após aumento da frequência de pacing para 75 bpm. No follow-up aos 3 meses teve apenas 4 episódios de taquicardia ventricular polimórfica não mantida, evidente na monitorização à distância (home monitoring) do CDI.
Foi conhecido o resultado do estudo genético, que revelou uma mutação c.1817C >T no codão 606 do gene KCNH2, em heterozigotia (Figura 5a). Esta troca do nucleótido leva à substituição do amino-ácido serina por uma fenilalanina (p.S606F) na proteína codificada pelo KCNH2. O aminoácido afectado localiza-se ao nível extracelular entre o 5.° e o 6.° domínios transmembranares da proteína. Encontrámos ainda no gene KCNH2 três outras variantes não patogénicas, já previamente descritas.
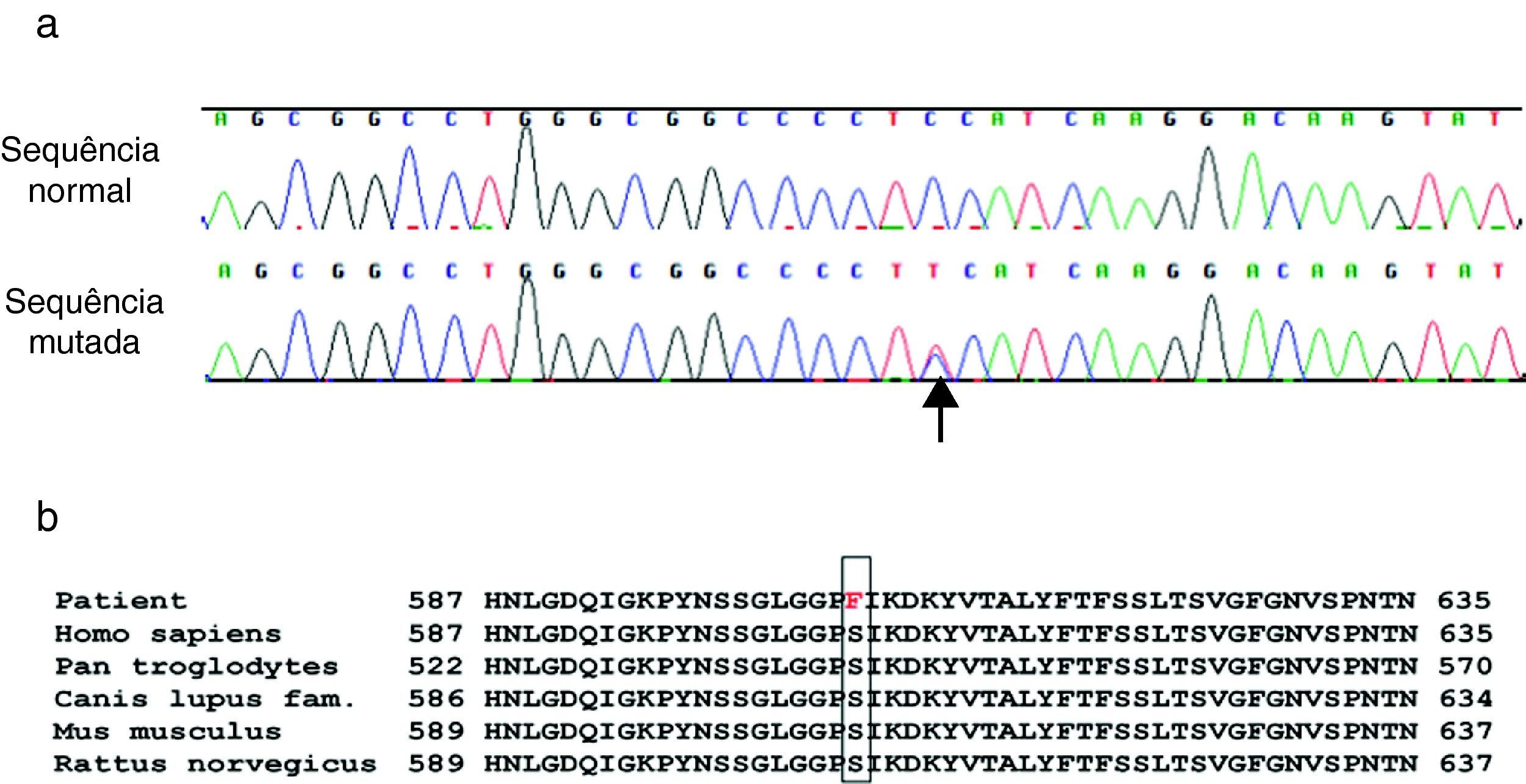
a) Mutação, em heterozigotia, c.1817 C>T p.S606F (seta), do gene KCNH2. A linha superior corresponde à sequenciação de um controlo normal. b) Homologia da sequenciação de aminoácidos em diferentes espécies testados com o programa NCBI HomoloGene (http://www.ncbi.nlm.nih.gov/homologene). O codão p.S606 mostra homologia nas diferentes espécies.
A sequênciação dos genes KCNQ1 e SCN5A não identificou alterações patogénicas.
A mutação c.1817C >T, levando a substituição de citosina por tiamina, ainda não foi descrita na literatura. O resíduo serina-606da proteína codificada pelo gene KCNH2 está altamente conservado em várias espécies (Figura 5b). A mutação p.S606F ainda não foi descrita na literatura, no entanto, esta mutação foi encontrada, por um grupo de investigadores de Oxford, UK, num doente e na sua mãe com «sintomas sugestivos de SQTL» (Melanie Proven, comunicação pessoal).
Estão em curso contactos para estudo clínico e genético dos familiares directos os quais permitirão o estudo da segregação da mutação.
DiscussãoA SQTL tem frequentemente uma apresentação que torna difícil o seu diagnóstico. Este assenta na história clínica e no ECG, sendo fundamental a análise do intervalo QTc1. No entanto, nem sempre é fácil efectuar o diagnóstico baseado no ECG, pois o prolongamento do intervalo QT pode não ser evidente em todas as derivações ou variar entre os vários ECGs3,7.
São frequentemente utilizados os critérios de diagnóstico de Schwartz (score de Schwartz), que inclui várias características eletrocardiográficas, para além do valor do intervalo QTc, como a morfologia da onda T, a evidência de bradicardia e torsade de pointes documentada; clínica de síncope, surdez congénita e a história familiar de morte súbita em jovens ou de SQTL em familiares directos7. Este score de probabilidade da doença, embora com grande especificidade tem baixa sensibilidade, dada a penetrância varíavel das mutações1, isto é, há portadores da mutação que não apresentam o fenótipo característico, mas que no entanto estão também em risco de arritmias ventriculares. Reveste-se pois, de extrema importância, a identificação genética e o estudo genético familiar para identificação de portadores da mutação.
Apesar do diagnóstico não ser ditado pelo estudo genético, esta avaliação possibilita a identificação dos genes e das mutações associadas, identificando a SQTL em doentes com intervalo QTc não diagnóstico e influenciando decisões terapêuticas1. As formas mais frequentes da SQTL apresentam mutações nos genes que codificam subunidades dos canais de potássio: KCNQ1 e KCNH2 (ou HERG, human ether a go-go), nos tipos 1 e 2, e nos canais de sódio: SCN5A, cujas mutações estão associadas ao tipo 3. As mutações do gene SCN5A estão também associadas à síndroma de Brugada1.
Como o caso descrito acima ilustra, o diagnóstico diferencial com epilepsia pode ser difícil e complexo. As convulsões na SQTL surgem como consequência dum período de hipoperfusão cerebral prolongado secundário à arritmia cardíaca. No entanto, Johnson e colaboradores mostraram recentemente, que o fenótipo convulsão está mais frequente associado à SQLT tipo 2, o que pode sugerir que as convulsões na SQTL tenham uma base fisiopatológica comum8. A epilepsia pode fazer parte desta síndroma, à semelhança da surdez na SQTL tipo 1 e da sintomatologia gastro-intestinal na SQTL tipo 38.
A base para esta evidência fenotípica pode ser explicada pelo facto da subunidade KCNH2 dos canais de potássio também ser expressa nas células da glia (astrócitos) do hipocampo. Estas regulam a concentração de potássio extra-neuronal, cuja modificação pode ser epileptogénica8.
É consensual que o limiar epiléptico se encontre diminuído secundariamente à hipoperfusão cerebral pela taquicardia ventricular polimórfica e à homeostasia do potássio extraneuronal no hipocampo. Alternativamente, é especulado que as convulsões, são de facto crises epilépticas secundárias às perturbações dos canais KCNH2 de potássio do hipocampo, correspondendo a uma epilepsia temporal8.
A favor desta hipótese está o facto de na epilepsia também encontrarmos síndromas com associação bem definida a mutações em canais de sódio e de potássio. São estas as convulsões neonatais familares benignas nos canais de potássio: KCNQ2 e KCNQ3, a epilepsia nas convulsões febris, associada aos canais de sódio SCN1B e SCN1A9.
A epilepsia também tem sido apontada como causa de arritmias malignas. Nashef e colaboradores levantaram a hipótese de que, nos doentes com epilepsia e morte súbita inesperada (SUDEP, sudden unexpected death in epilepsy), a morte seja causada por depressão cardio-respiratória central durante a crise, que conduz a uma predisposição a arritmias ventriculares e morte10.
Na nossa doente nunca foi documentada actividade epiléptica, mesmo durante os episódios sincopais disrítmicos. Aproximadamente 10-40% dos doentes com epilepsia não mostram actividade epiléptica no EEG pelo que um EEG normal ou inespecífico não exclui o diagnóstico de epilepsia11. O diagnóstico diferencial entre a SQTL e a epilepsia pode ser um desafio, mas é fundamental. A instituição de terapêutica anti-epiléptica pode, por inibição da subunidade codificada pelo gene KCNH2, conduzir ao prolongamento do intervalo QT e aumentar a susceptibilidade para taquicardia ventricular polimórfica. Numerosos fármacos apresentam este efeito, nomeadamente o fenobarbital que era administrado a esta doente12.
A SQTL deve ser considerada em indivíduos jovens com convulsões e EEG que não exclua epilepsia. Estudos mostram que um terço das «epilepsias resistentes» podem ser variantes da SQTL13. Desta forma destaca-se o papel do ECG de Holter de 24 horas e o vídeo-electroencefalograma com ECG, que podem aumentar exponencialmente a acuidade diagnóstica, nomeadamente se os registos incluírem o evento arrítmico, tal como se verificou neste caso.
No entanto, há a destacar as características clínicas da síncope da SQTL como ponto fulcral para o seu diagnóstico, uma vez que a arritmia surge na presença de um factor desencadeante, um stress fisiológico, que é específico de mutação. O tipo 1 ocorre mais frequentemente em associação a stress físico e caracteristicamente com a natação/mergulho; o tipo 2 ocorre em associação a sons intensos súbitos e emoções, tal como verificado na doente apresentada; no tipo 3 a arritmia ocorre sem o factor emoção em repouso ou durante o sono e sem o despertar do doente1,8.
O diagnóstico e a terapêutica adequada destes doentes, podem impedir os eventos fatais que caracterizam esta síndrome. A MS na SQTL ocorre em 1 a 2% por ano, 20% no primeiro ano de diagnóstico14. A instituição de terapêutica beta-bloqueante reduz o número de eventos e morte súbita cardíaca em 70%15, pelo que é recomendada nos doentes com este diagnóstico clínico5. Mais de 10% podem apresentar paragem cardíaca e MS apesar do tratamento15, sendo mandatória a implantação de CDI nos doentes sobreviventes a paragem cardíaca5.
O risco arritmogénico não é igual nos três tipos da SQTL1,5. É mais elevado nas SQTL tipo 2 e 3, pelo que o genótipo é tido em conta nas indicações para implantação de CDI5. A eficácia da terapêutica beta-bloqueante também não é igual nos diferentes tipos da SQTL, sendo menor no SLQT tipo 315. Realça-se assim a necessidade da identificação da mutação específica.
Neste caso houve a identificação de uma nova mutação p.S606F, não previamente descrita, no gene KCNH2 (HERG), da SQTL tipo 2. As localizaçõs da mutação no domínio transmembranar e o facto de codificar um aminoácido altamente conservado nas espécies, confere características de patogenecidade à mutação.
A simpaticectomia cardíaca esquerda surge como opção terapêutica na SQTL, face à falência dos beta-bloqueantes. A redução da estimulação adrenérgica, enquanto trigger, e a modificação do substrato arritmogénico, evidente na redução do intervalo QTc, são responsáveis pela diminuição de eventos cardíacos16 aos 3 meses de follow-up. No entanto, o intervalo QTc de 502ms pós-simpaticectomia confere menor probabilidade de bom prognóstico, mantendo-se associado a elevada probabilidade de arritmia e MS16, tal como já era anteriormente determinado pelo genótipo da SLQT tipo 2 e pela gravidade clínica prévia à cirurgia.
A existência de uma tempestade arrítmica em doentes portadores de CDI constitui uma complicação grave. A exclusão de factores que potenciam o prolongamento do intervalo QT é fundamental. A febre, reconhecida como factor desencadeante de arritmia na Síndroma de Brugada, foi apenas descrita pontualmente na SQTL condicionando prolongamento do QTc e em associação com uma mutação específica (A558P missense mutation no HERG)17. Neste caso embora os períodos de arritmia fossem documentados durante período febril, o intervalo QTc não estava mais prolongado do que o basal da doente (QTc=462ms).
Por outro lado, o facto da tempestade arrítmica ter surgido no contexto de infecção a H1N1, levanta a hipótese da existência de um subgrupo de doentes com SQTL susceptível a tempestade arrítmica na presença desta infecção viral. Este subgrupo de alto risco pode apresentar um novo mecanismo arritmogénico associado especificamente à nova mutação identificada e à infecção a H1N1.
ConclusãoDestaca-se a SQTL enquanto etiologia de síncope e de crise convulsiva, reforçando-se a necessidade do diagnóstico diferencial com epilepsia. No entanto, no actual estado da arte, está por definir se existe ou não um mecanismo molecular que determine uma ligação entre a SQTL e a epilepsia.
Uma nova mutação no gene KCNH2 foi identificada e pela primeira vez descrita. Esta mutação parece conferir susceptibilidade a tempestade arrítmica, no contexto de síndromes febris/virais.
Conflito de interessesOs autores declaram não haver conflito de interesses.
