




Neurofibromatosis type 1 (NF1) is a common autosomal dominant genetic disorder that affects multiple organ systems and has a wide range of clinical manifestations. Pulmonary hypertension (PH) associated with NF1 (PH-NF1) is rarely seen, but confers a dismal prognosis. In the literature this association has been described in only 31 cases. The authors report the case of a 77-year-old female patient with NF1 complicated by severe precapillary PH despite triple disease-specific oral combination therapy. Because no data are available on the efficacy of specific PH therapy in PH-NF1, these patients should be assessed in expert PH centers and referred for lung transplantation at an early stage.
A neurofibromatose tipo 1 (NF1) é uma doença genética frequente com transmissão autossómica dominante, que afeta diversos órgãos e apresenta uma grande variedade de manifestações clínicas. A associação entre a hipertensão pulmonar (HTP) e a NF1 (HTP-NF1) é rara, apresentando, no entanto, um mau prognóstico. Na literatura esta associação encontra-se descrita em apenas 31 casos. Os autores descrevem o caso de uma mulher de 77 anos com NF1 complicada por HTP pré-capilar grave a pesar de a mesma estar sob terapêutica tripla específica para HTP. Como não existe evidência sobre a eficácia da terapêutica específica na HTP-NF1 estes doentes devem ser avaliados em centros especializados em HTP e referenciados para o transplante pulmonar precocemente.
Neurofibromatosis type I (NF1) or von Recklinghausen disease is a genetic disease with an incidence of approximately 1 per 3500 individuals. It is transmitted as an autosomal dominant and fully penetrant trait.1 NF1 is caused by mutations in NF1, the gene coding for neurofibromin 1, a negative regulator of the Ras signal transduction pathway that has a role in both tumor suppression and regulation of cell growth and proliferation.2 NF1 is characterized by prominent skin features (hyperpigmented macules termed café-au-lait spots and neurofibromas), optic tumors and other central nervous system tumors, bony abnormalities, learning disabilities and an increased risk of certain non-nervous system cancers.3
NF1 is a multisystem disease and lung disease, especially pulmonary hypertension (PH), can be one of its most severe complications. PH associated with NF1 (PH-NF1) is a rare but severe complication of NF1.
In a recent literature review, Jutant et al.4 identified 18 articles describing 31 cases of NF1 associated with precapillary PH (defined by a mean pulmonary artery pressure ≥25 mmHg and pulmonary artery wedge pressure <15 mmHg) measured by right heart catheterization. PH-NF1 is classified as Group 5 PH, defined as ‘PH with unclear and/or multifactorial mechanisms’, a heterogeneous group including several disorders with multiple mechanisms, for which there are neither data nor recommendations regarding the use of drugs approved for pulmonary arterial hypertension (PAH).5 PH-NF1 confers a dismal prognosis, and these patients should be assessed in expert PH centers and referred for lung transplantation at an early stage.4
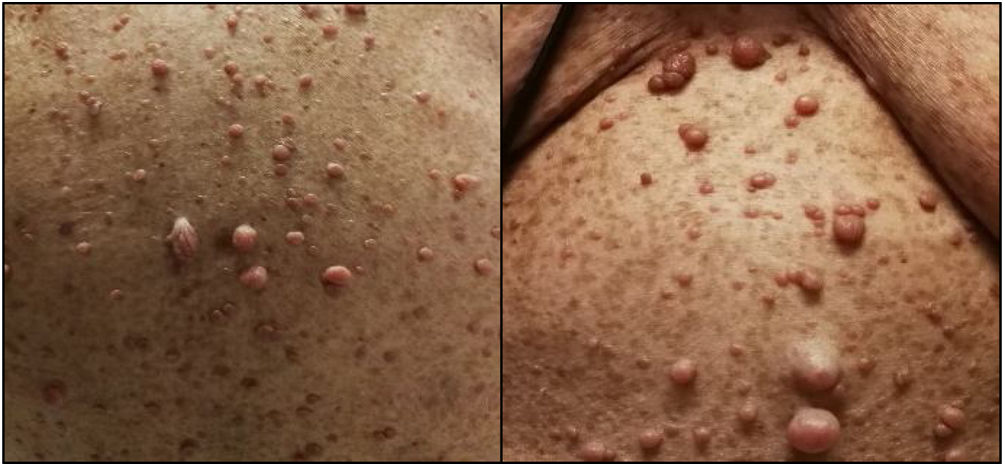
Case reportA 77-year-old female patient with a history of NF1 was referred to the cardiology clinic because of a history of worsening dyspnea over the previous 18 months. On assessment she had tell-tale signs of NF1, namely multiple café-au-lait spots, axillary freckling and neurofibromas all over the body (Figure 1). She was in New York Heart Association (NYHA) functional class III at the first medical visit. The electrocardiogram showed right-axis deviation, right atrial enlargement and right ventricular strain (Figure 2A). The chest X-ray revealed a prominent central pulmonary artery with peripheral pruning. Echocardiography (Figure 2B) showed enlarged right-side chambers, right ventricular hypertrophy, flattening of the interventricular septum and D-shaped left ventricle (LV). There was also mild tricuspid regurgitation with velocity of 4.01 m/s and severe PAH with pulmonary artery systolic pressure (PASP) 72 mmHg. High-resolution computed tomography was remarkable for markedly enlarged central pulmonary arteries and bilateral mosaic pattern of ground glass haziness (Figure 2C). Pulmonary function tests showed no alterations, however the patient did not tolerate the carbon monoxide diffusing capacity test. Right heart catheterization confirmed the presence of precapillary PH and absence of acute vasoreactivity response. We failed to identify risk factors for pulmonary artery hypertension (PAH), such as anorexigen use, portal hypertension, HIV infection, connective tissue diseases, congenital heart disease, drug and toxin exposure, hemoglobinopathies, chronic lung disease or thromboembolism. At the first medical visit tadalafil 20 mg twice daily was initiated. Reassessment at four months revealed no improvement in pulmonary pressures or symptoms and macicentan 10 mg once daily was added. Within a year the patient was hospitalized twice for decompensated heart failure (brain natriuretic peptide >3000 pg/ml) and at this point long-term oxygen therapy and diuretics were started. A recent transthoracic echocardiogram demonstrated PASP >70 mmHg and specific triple therapy with selexipag 200 mg twice daily was recommended. Despite triple therapy, at the last visit she was in NYHA class IV.
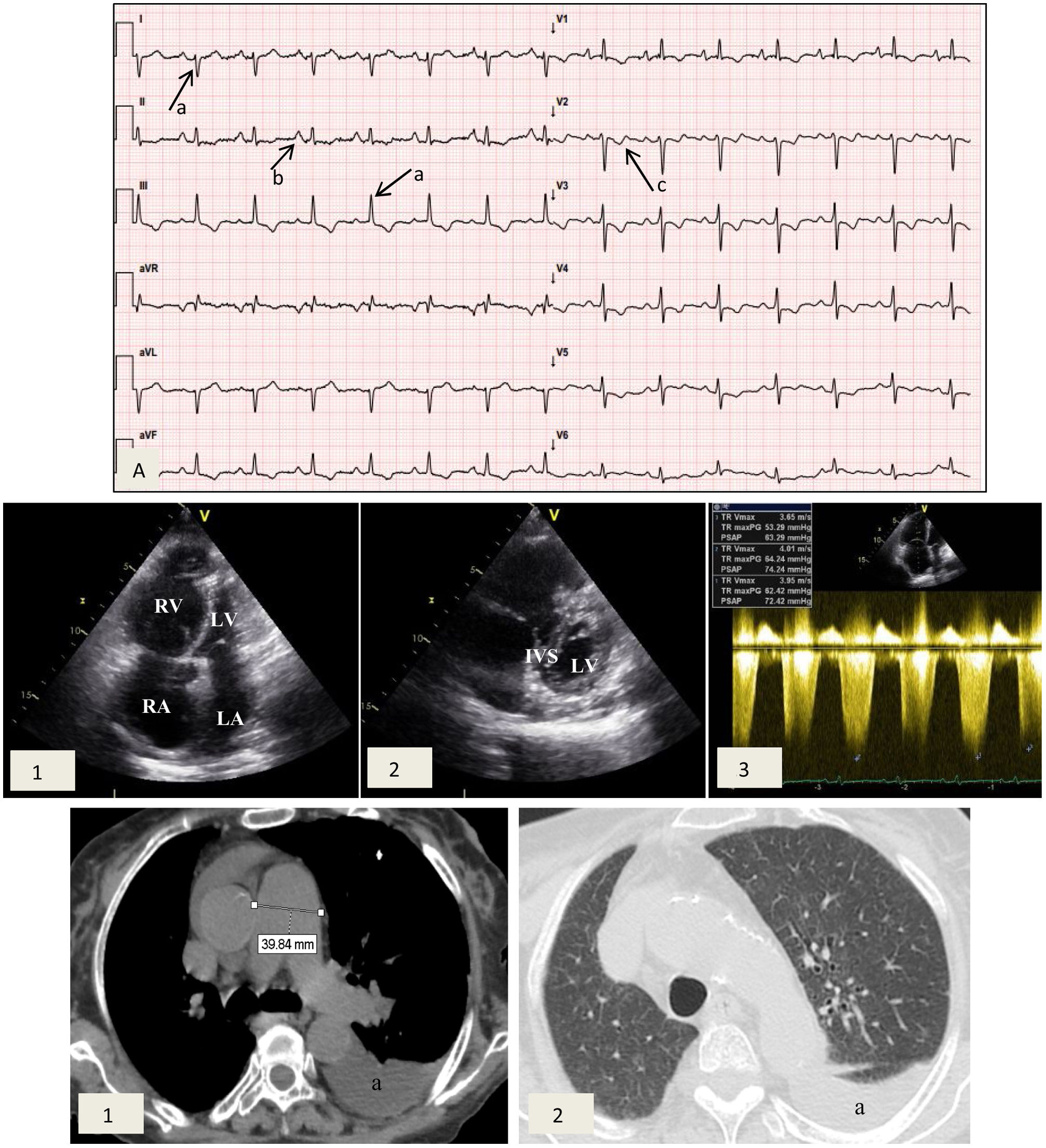
Cardiac studies obtained during medical visits. (A) Electrocardiogram showing right axis deviation >120° (a), right atrial enlargement (b) and right ventricular strain (c); (B) transthoracic echocardiogram depicting (1) enlarged right atrium and right ventricular dilation and hypertrophy and (2) flattening of the interventricular septum and D-shaped left ventricle; (3) pulmonary artery systolic pressure 72 mmHg. IVS: interventricular septum; LA: left atrium; LV: left ventricle; RA: right atrium, RV: right ventricle; (C) high-resolution computed tomography exhibiting (1) enlarged central pulmonary arteries and (2) bilateral mosaic pattern of ground glass haziness. Left pleural effusion is also visible (a).
Herein we report the case of a patient with PH-NF1. NF1, a common autosomal dominant genetic disorder, presents a fully penetrant trait in adults, but many manifestations of the disease increase in frequency or severity with age.6
The diagnosis of NF1 is clinical, based on criteria proposed by the National Institutes of Health Consensus Development Conference.7 If two or more of the seven criteria are present in the same patient, the diagnosis is established. The present patient fulfilled the criteria for diagnosis of NF1 because she had (1) six or more café-au-lait macules (>1.5 cm), (2) two or more cutaneous neurofibromas and (3) axillary or groin freckling. The average life expectancy of individuals with NF1 is reduced by 10-15 years and the most common cause of early death is cancer.8–10
NF1 is caused by mutations of the NF1 gene, located at chromosome 17q11.2,11 which codes for neurofibromin 1, a cytoplasmic protein that has a role in tumor suppression.1 Inactivation of the gene through mutation leads to a loss of neurofibromin and to a constitutive activation of Ras pathways,12–14 leading to deregulation of cell proliferation and differentiation and to the development of benign neurofibroma-like tumors and malignant peripheral nerve sheath tumors, and probably also to the lung complications seen in NF1.4
The features of NF1 are extremely varied and vasculopathy is a recognized but rare expression of the disease.7 Characteristically there is a proliferation of intima and media layers usually involving renal, mesenteric and aortic vessels.7,15 Involvement of pulmonary arterial beds and pulmonary hypertension is unusual in NF1.7,15,16
The 31 cases in the literature review by Jutant et al.4 revealed few characteristic features of PH-NF1. Although NF1 affects male and female patients without gender predominance, there is a female preponderance in PH-NF1 (male:female ratio 1:4.24), suggesting that estrogen plays a role, as reported in heritable PAH.17
In contrast with heritable or idiopathic PAH,18 PH occurs late in the course of NF1, with a median age at diagnosis of 57 years (50-65 years).4 In the present case PH was found even later, at 77 years. This suggests that arteriopathy of the pulmonary vasculature is a late phenomenon in the natural history of NF1 and presents indolent symptoms. Besides, there is a lack of awareness of this association and the diagnosis is consequently often delayed.
Similar to this case, dyspnea and signs of right heart failure lead to assessment for PH. In Jutant et al., at diagnosis 75% of the patients were in NYHA functional class III or IV and most had severe hemodynamic impairment and showed a negative vasoreactivity test, which is associated with lack of response to calcium channel blockers.4
Zamora et al.19 reported that NF1 may be associated with parenchymal lung involvement in adulthood, characterized by lung cysts or bullae in the upper lobes, diffuse ground-glass opacities and reticular opacities, sometimes with pathologic evidence of fibrosis. In our case lung involvement was manifested by ground-glass opacities. However, in Jutant et al.’s review, one third of PH-NF1 cases presented with no lung parenchymal disease, suggesting specifically vascular lesions.4
Sometimes NF1-PH patients have impaired diffusion capacity but other parameters are normal, suggesting vascular involvement. By contrast, NF1 patients with lung disease show some obstructive, restrictive or mixed patters and male preponderance.19
Use of PAH-specific oral therapy in NF1 patients has had discouraging results. An aggressive therapeutic approach, based on a phosphodiesterase-5 inhibitor and an endothelin receptor antagonist, is currently advocated.20 Even so, a progressive course with development of cor pulmonale and terminal right cardiac failure is frequent.16,21 In the presence of mild PH with severe lung disease, symptomatic treatment with long-term oxygen therapy, diuretics and noninvasive ventilation has been proposed.4 In cases of severe PH without extensive lung involvement PAH-specific therapy should be discussed. The risk/benefit ratio of these treatments in PH-NF1 is unknown, because of the risk of pulmonary edema associated with venous and capillary involvement.22 For this reason patients need to be referred early to expert PH centers and considered for lung transplantation.4
Giannakoulas et al.23 reported the successful management of a PH-NF1 patient with atrial septostomy in conjunction with PAH-targeted therapy. The creation of a shunt, by decreasing right ventricular preload and increasing left ventricular preload, with consequent increase in cardiac output, facilitated the initiation and uptitration of prostanoid infusion. Treatment with sorafenib, a tyrosine kinase inhibitor, was initiated in one patient, who experienced mild clinical and hemodynamic improvement after three months, but no data on long-term response was available.24
Given the known effects of neurofibromin on growth regulation signaling, several currently available agents, in particular drugs that inhibit the dysregulation induced by neurofibromin deficiency, are potentially targets in PH-NF1.25 Nevertheless, no treatments have been evaluated for this particular indication.
Our patient did not improve with triple therapy, and addition of another therapeutic class or atrial septostomy should be discussed at the next visit. Lung transplantation is no longer an option for this patient.
ConclusionPH is a rare but potentially severe complication of NF1. Only 31 cases have previously been reported and we describe one more. The mechanisms of PH-NF1, classified as group 5 PH, ‘PH with unclear and/or multifactorial mechanisms’, remain poorly understood and may include lung parenchymal involvement and pulmonary vascular remodeling. PH-NF1 is characterized by late onset, female predominance and severe functional and hemodynamic impairment.
There are no guidelines regarding the treatment of PH in these patients, however several promising treatments targeting the Ras pathways should be studied. Because this is a rapidly progressive disease, patients should be referred early to expert PH centers and considered for lung transplantation.
Conflicts of interestThe authors have no conflicts of interest to declare.
