



Hypertrophic cardiomyopathy (HCM) is known as the most common genetic heart disease, characterized by otherwise unexplained left ventricular (LV) hypertrophy. In spite of major advances in whole genome sequence techniques, it is still not possible to identify the causal mutation in approximately half of HCM patients. Consequently, a new HCM concept, “beyond the sarcomere” is being developed, supported by data from recent HCM registries which reveal two distinct HCM subgroups: sarcomere positive HCM subgroup and nonfamilial HCM subgroup. Sarcomere positive HCM patients tend to be younger age at diagnosis, have fewer co-morbidities, present more often with reverse septal morphology, more myocardial fibrosis, less LV outflow tract obstruction, and a worse prognosis when compared to nonfamilial HCM patients. These subgroups, with different molecular basis, phenotypes and clinical profiles, will likely require specific management strategies.
Important research advances have also been made concerning diagnosis, sudden cardiac death stratification and therapy. In this article, we seek to review recent relevant knowledge, summarizing the advances in this complex and heterogeneous disease.
A miocardiopatia hipertrófica (HCM) é conhecida como a doença cardíaca genética mais frequente, caracterizada pela presença de hipertrofia ventricular esquerda desproporcional às condições de carga. Apesar dos grandes avanços nas técnicas de sequenciação do genoma completo, ainda não é possível identificar a mutação causal em cerca de metade dos doentes com HCM. Consequentemente, um novo conceito de HCM, «para lá do sarcómero» encontra-se em desenvolvimento, apoiado em dados de registos de HCM recentes que evidenciam dois subgrupos distintos de HCM: subgrupo HCM mutação sarcomérica positiva e subgrupo HCM não familiar. Doentes com HCM mutação sarcomérica positiva tendem a apresentar idade mais jovem à data do diagnóstico, menos comorbilidades, mais frequentemente morfologia septal reversa, mais fibrose miocárdica, menor obstrução da câmara de saída do ventrículo esquerdo e pior prognóstico quando comparados com os doentes com HCM não familiar. Esses subgrupos, com bases moleculares, fenótipos e perfis clínicos distintos, provavelmente necessitarão de estratégias de prevenção e tratamento específicas.
A investigação em HCM tem feito igualmente grandes progressos na última década relativamente ao diagnóstico, estratificação do risco de morte súbita e terapêutica farmacológica e não farmacológica. Neste artigo tentaremos rever e resumir os avanços mais relevantes no conhecimento desta complexa e heterogénea patologia.
Hypertrophic cardiomyopathy has been known as the most common genetic heart disease, with an initially estimated prevalence of 1:500 in the general population,1 whereas more recent studies indicate a prevalence of 1:200.2 Clinical diagnosis of HCM is based on unexplained left ventricular (LV) hypertrophy, which may present in a wide variety of patterns.3 Similarly, it is a disease with significant clinical heterogeneity, including patients who remain asymptomatic, reaching longevity equivalent to the general population,4 but, on the other hand, it is one of the main causes of sudden death in young people5 and athletes.6 Between these two extremes on the clinical spectrum, there are several evolution profiles, with very different clinical, therapeutic and prognostic implications: 1. heart failure (HF) profile, either due to diastolic dysfunction and/or obstruction, or systolic dysfunction; 2. atrial fibrillation (AF) and stroke profile; 3. sudden cardiac death (SCD) profile.3,4,7,8
Molecular basisHypertrophic cardiomyopathy has been considered predominantly an autosomal dominant (AD) genetic disease, although some cases are explained by de novo mutations and, less frequently, by autosomal recessive heredity.9 It is known as a sarcomere disease, and its pathogenic variants are described in almost all sarcomere proteins; however, 70% of the identified mutations are in the myosin heavy chain genes and myosin-binding protein C. All other genes involved, including troponin T, troponin I, alpha-tropomyosin, alpha actin, myosin light chain, present a much lower frequency of pathogenic variants and some are “private” mutations, present in only one family. In conventional genetic tests, disease-causing mutations are screened using next generation sequencing panel on at least eight key sarcomeric genes. Additional genes whose mutations can mimic HCM are also investigated to detect HCM phenocopies (e.g. Fabry's disease or Danon syndrome), with very different therapies and prognosis. The diagnostic power of these tests is modest, approximately 46% in the HCM patient population,10 especially based on the assumption that HCM is a genetic disease mainly with AD transmission.
In a recent study, Bagnall et al,11 studied the incremental value of whole genome sequencing (WGS) over conventional genetic tests in a cohort of 58 HCM probands. The authors detected a pathogenic or probably pathogenic variant in 20% of families with inconclusive prior conventional genetic testing. These detected cases consisted mainly of variants in genes not included in the previous genetic test panel and pathogenic variants in non-coding regions, including four deep intronic variants that activate cryptic splicing and a variant in the mitochondrial genome. Cryptic splice-altering variants are, in fact, now an emerging cause of HCM, documented in other studies.12,13 Even so, as an initial approach, in families who had not undergone prior conventional genetic testing, WGS presented a diagnostic power of 42% in the former study.
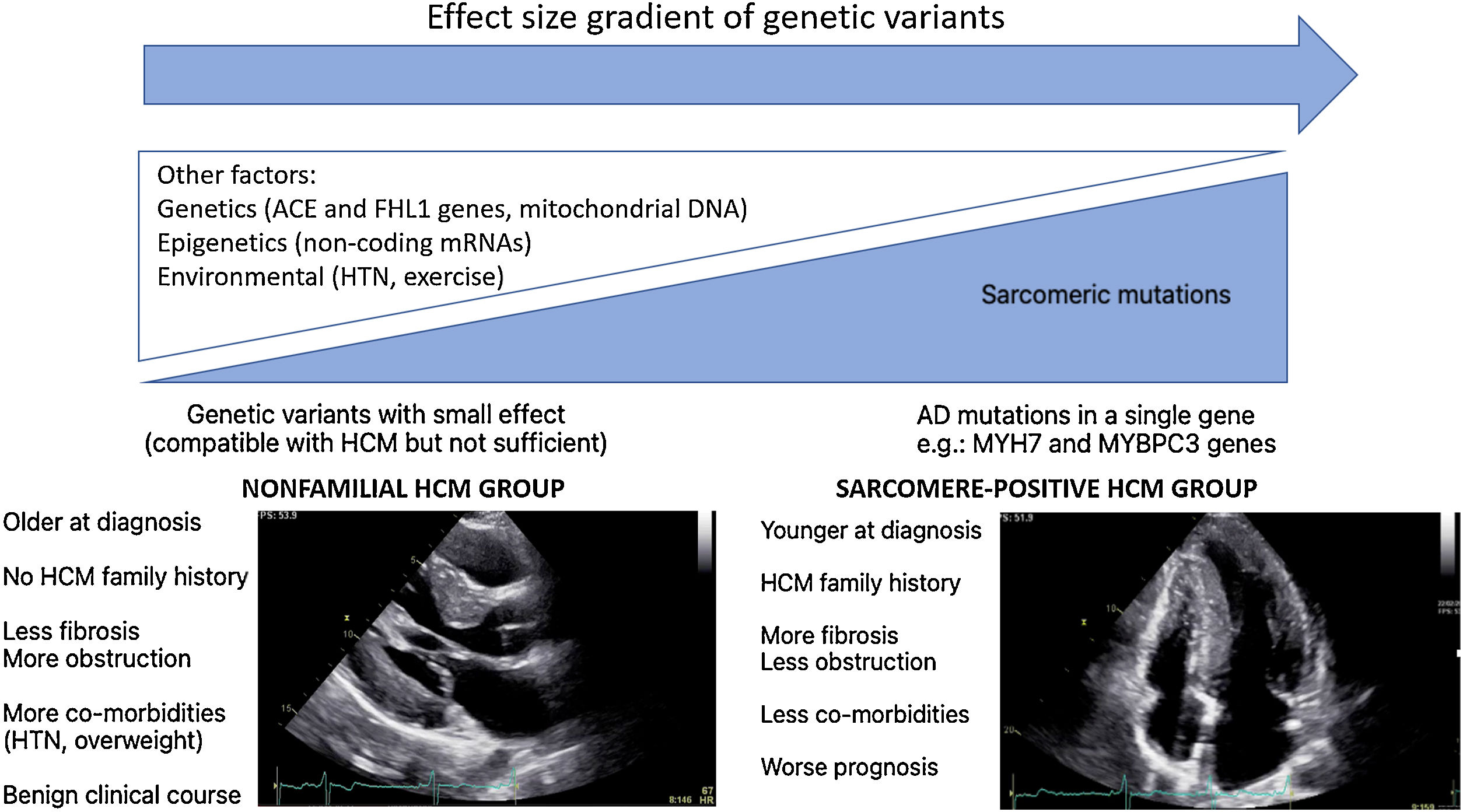
In WGS era, one would expect a significant increase in the identification of HCM genetic basis. This “missing causal gene” in HCM may be due to the difficulty in attributing causality in multiple genetic variants, in an unambiguous way, especially in sporadic cases and in small families.14 Another explanation may lie in the “effect size” gradient of genetic variants (Figure 1). It may be necessary to rethink the HCM hypothesis paradigm as an exclusively genetic and monogenic disease, in favor of a model “beyond the sarcomere”,15 in which, like other pathologies such as dilated cardiomyopathy, there is a genetic substrate, which is influenced by many other factors: genetics, epigenetics or even environmental factors. This model would explain the difficulty in stablishing predictable genotype-phenotype correlations for HCM causing mutations. In fact, in recent years several genotype-phenotype associations have emerged (Table 1), yet the variability among family members with the same mutation but different phenotypes remains unexplained. This model would also explain the cases of HCM with negative genetic testing, without the classic AD mutations required to cause disease. These nevertheless present a genetic substrate compatible with HCM, or perhaps with pathogenic variants with an intermediate or even clinically indiscernible effect, but which, in the appropriate context and influenced by other factors involved in cardiac hypertrophy and fibrosis regulation, can promote HCM phenotype expression (Figure 1).
Emerging genotype-phenotype associations.
| Gene | Phenotype | Comment | Reference |
|---|---|---|---|
| Thin filament gene mutations (TNNT2, TNNI3, TPM1 and ACTC) | • Milder and more atypical hypertrophy (concentric and apical patterns)• Increased LV fibrosis• Higher rate of HF progression• Higher likelihood of adverse remodeling leading to systolic or severe diastolic disfunction• Similar rates of ventricular arrhythmias and SCD | vs. thick-filament gene mutations | 58 |
| TNNI3 mutations | • Restrictive phenotype | 59 | |
| ACTC mutations | • Apical hypertrophy | 60 | |
| MYH7 mutations | • Higher rate of incident AF (independent of clinical and echocardiographic factors) | vs. other sarcomeric gene mutations | 61 |
| • Earlier age at diagnosis | vs. no sarcomere mutation | 20 | |
| • More frequent evolution to heart transplant | vs. MYBPC3 mutations | 20 | |
| • More ventricular arrhythmias | vs. MYBPC3 mutations or no sarcomere mutation | 20 | |
| • More cardiac conduction disease | vs. MYBPC3 mutations | 20 | |
| • Restrictive phenotype | 59 | ||
| MYBPC3 mutations | • Elderly onset and lower penetrance | vs. MYH7 mutations | 9 |
| PLN | • NSVT | 18 |
Abbreviations: AF: atrial fibrillation; ACTC: cardiac alpha actin gene; ANK2: ankyrin B gene; HF: heart failure; LV: left ventricle; NSVT: non-sustained ventricular tachycardia; PLN: phospholamban gene; SCD: sudden cardiac death; SCN5: Sodium Voltage-Gated Channel Alpha Subunit 5 gene, TNNT2: cardiac troponin T gene; TNNI3: cardiac troponin I gene; TPM1: cardiac a tropomyosin gene. Emerging genotype-phenotype correlations, including sarcomeric variants (MYH7, TNNT2, TNNI3, TPM1 and ACTC) and the modifier effect of non sarcomeric variants (ANK2, SCN5, PLN).
There is, in fact, a growing body of evidence suggesting two subgroups of HCM, based on genetic testing, presenting different phenotypes.16–19 Sarcomere positive (SARC+) HCM patients are younger at diagnosis, present a higher degree of hypertrophy, typically asymmetrical, and have a more frequent family history of HCM or SCD.16–19 In contrast, sarcomere negative (SARC-) HCM patients have comorbidities more frequently, such as hypertension (HTN)18,19 and being overweight19 and follow a much more benign clinical course.18,19 Notably, several studies have associated SARC+ status with higher rates of cardiovascular death18,19and SCD.18,20,21
Regarding the Portuguese context, the Portuguese HCM Registry22 reports globally a low percentage of familiar HCM, a high percentage of cases diagnosed after >65 and a low global risk profile, which supports the hypothesis that, in the real world, the nonfamilial subgroup with a more benign prognosis predominates. In an ancillary study of the aforementioned registry focusing on genetic characterization and genotype-phenotype associations,21 again SARC+ patients were younger, more often presenting family history of HCM and SCD, revealing a higher prevalence of asymmetrical hypertrophy and a higher association with SCD.
Ongoing and recent registries also support this idea. The HCMR international registry,23 currently underway and involving 2755 genotyped patients, characterized two groups of patients at baseline: 1. SARC+ group, in which reverse septal morphology was more frequent and associated with more fibrosis and less obstruction at rest; 2. SARC- group, in whom the sigmoid septum morphology predominated, with less fibrosis but more obstruction. The recently published SHaRe international registry,10 which involved 4591 HCM patients with an average follow-up of 5.4 years and >24 000 patient-years, identified two predictors of adverse outcomes: sarcomeric mutation status and age at diagnosis. Survival analysis showed that groups with 1. pathogenic or likely pathogenic mutation and 2. variants of unknown significance (VUS) had an earlier onset of events and a higher incidence of composite outcome (HF and AF) when compared to the SARC- group. Regarding malignant ventricular arrhythmias endpoint, the group with pathogenic or likely pathogenic mutation had a higher risk than the group without any identified mutation. Conversely, patients with nonfamilial HCM (negative mutations and without family history of HCM) had a lower risk of mortality and composite outcomes when compared to other HCM patients. The age-adjusted mortality of this group was similar to that of the general population.
Regarding age, mortality in young HCM patients (20–29 years) was four times higher and in patients between 50–69 years it was three times higher than that of the general population of the same age group, respectively. SCD was not the main cause of mortality in these patients, occurring in only 16% of deaths. Main causes of mortality were HF and non-cardiac mortality.
Regardless of age at diagnosis, most complications related to HCM occurred late in life, between 50–70 years old, and the main events were AF and HF. Cumulative lifetime incidence of malignant ventricular arrhythmias was 32% in patients whose age at diagnosis was <40 years but was rarely found in the group with a diagnosis >60 years (1–2%). Although patients diagnosed at younger ages are more likely to experience events throughout life, time amortizes the probability of having an event. Therefore patients diagnosed at older age, despite having a lower cumulative lifetime incidence, have a higher annual incidence of events when compared to young patients.
Overall, these findings suggest: 1. Genetic study may indeed play a role in stratifying the prognosis of HCM patients; 2. VUS may have an impact on pathophysiological mechanisms (and may include some variants that are actually pathogenic but yet to be demonstrated) and in prognostic stratification, probably identifying an intermediate risk class; 3. Nonfamilial HCM may be a subgroup of disease very different from familiar HCM; probable prevention, treatment of these patients and screening of relatives should also be very different in these two groups; 4. HCM was shown to be a disease with significant morbidity and mortality throughout its evolution, mainly due to the AF and HF outcomes despite contemporary therapies at reference centers, highlighting the need for lifelong follow-up.
DiagnosisIn adults, HCM is defined as LV wall thickness ≥15 mm in one or more myocardial segments, which is not exclusively explained by loading conditions and occurrs in the absence of another cardiac or systemic, metabolic disease or in the context multiorgan syndrome associated with LV hypertrophy.3 Transthoracic echocardiography is the first imaging modality3 and establishes the diagnosis in the majority of cases. Additionally, cardiac magnetic resonance (CMR) should be considered at baseline assessment if local resources and expertise permit.4 CMR, the gold standard imaging technique in morphology and wall thickness assessment, may also detect focal or diffuse fibrosis through late gadolinium enhancement (LGE) and T1 mapping respectively, which, when present, may support HCM diagnosis in borderline cases. Interestingly, extracellular volume (ECV) and native T1 mapping appear to differentiate HCM from HTN24 and sarcomere mutation carriers from controls,25 even in the absence of LV hypertrophy or LGE.
Diagnosis of HCM is currently based on only one of the characteristics of the disease, the increased LV wall thickness, when in fact the disease also involves myocardial disarray, interstitial fibrosis, microvascular remodeling and microcirculatory dysfunction. Emerging imaging techniques, addressing virtual histology, may offer non-invasive alternatives enabling a more accurate diagnosis of HCM in the future. Shear wave elastography (SWE), a non-invasive technique derived from echo that quantitatively evaluates the stiffness of the tissues,26 is a promising tool in the evaluation of patients with HCM, in which the development of myocardial disarray, microvascular remodeling and interstitial fibrosis is associated with increased myocardial stiffness, diastolic dysfunction and disease progression. Recent data have documented the increase in myocardial stiffness assessed by SWE in patients with HCM when compared to healthy volunteers.26 This new tool, apparently less dependent on loading conditions than conventional diastolic function assessment parameters, can be useful in the early detection of myocardial structural changes and in monitoring the response to therapy.
Cardiac magnetic resonance may also improve tissue characterization in HCM, including through techniques derived from cardiac diffusion tensor imaging such as fractional anisotropy (FA). This technique assesses the orientation of myofibrils, and is therefore able to quantify the degree of myocardial disarray and assist in the diagnosis and characterization of myopathy in patients with HCM.27 A recent study by Ariga et al.28 reported a reduction in diastolic FA in HCM patients when compared to healthy controls. LGE and ECV were significant predictors of FA, in line with the fact that fibrosis also decreases FA. However, FA adjusted for LGE and ECV remained reduced in HCM patients, probably accounting for myocardial disarray without fibrosis. Additionally, an association between a reduction in FA and ventricular arrhythmias was detected, which remained significant after correcting for LGE, ECV and wall thickness. These promising techniques are still far from being part of routine clinical practice, so further developments are awaited.
Although classic clinical diagnosis of HCM is based on imaging techniques, in recent decades, the role of genetic diagnosis has increased.4 Indeed, genetic testing in HCM is useful for confirming diagnosis, preclinical diagnosis, cascade genetic testing in the family and guiding reproductive decisions.29 However, current guidelines recommend genetic testing mainly when familiar genetic screening is anticipated or when clinical presentation suggests a specific and non-sarcomeric genetic etiology, such as Anderson-Fabry disease, Danon disease or familial amyloidosis.30 Genetic testing in patients with an equivocal clinical diagnosis such as borderline hypertrophy and concomitant HT, valve disease or in athletes should only be performed by experienced teams after an exhaustive clinical and family evaluation,4 since the result can also be difficult to interpret.4,30
Clinical profile and prognosisWhat is new in sudden cardiac death risk stratificationIn the past decade, HCM SCD risk stratification has been an area of intense research and discussion. In 2011, American guidelines7 recommended implantable cardioverter defibrillator (ICD) implantation for primary prevention (Class IIa recommendation) in patients with family history of SCD in a first-degree relative, with severe hypertrophy (≥30 mm) or who presented one or more episodes of recent and unexplained syncope. In patients with non-sustained ventricular tachycardia (NSVT) or an abnormal blood pressure response to exercise (ABPRE), ICD placement could be considered (Class IIb recommendation) in the presence of other SCD modifying risk factors.
In 2012, O’Mahony et al.31 performed a validation study of this strategy concluding that it is effective in preventing SCD in HCM patients, but its low discriminatory power results in the implantation of ICDs in a much larger number of patients than really needed. In 2014, a new SCD risk prediction clinical model was published,5 using mathematical models to weigh up the effects of specific risk markers, which are assessed as continuous variables, as opposed to the American strategy which treats risk factors, such as LV wall thickness, as binary variables. The HCM Risk-SCD model was included in the 2014 European HCM guidelines4 and is accessible as an online calculator. By entering the values obtained for seven clinical and imaging parameters, it is possible to obtain 5-year SCD risk estimate for a specific patient (Table 2).
European and 2020 American College of Cardiology/American Heart Association strategies for primary prevention of sudden cardiac death in hypertrophic cardiomyopathy patients.
| Risk factor | European HCM Risk-SCD model | 2020AHA/ACC strategy |
|---|---|---|
| Age | √ (variable included in a continuous way) | √ (although the decision to ICD implantation in patients older than 60 years should be individualized) |
| Maximum LV wall thickness | √ (variable included in a continuous way) | √ (variable included in a dichotomously way)(class of recommendation 2a) |
| Left atrial size | √ (variable included in a continuous way) | × |
| Maximum LVOT gradient | √ (variable included in a continuous way, determined at rest and with Valsalva maneuver, independent of concurrent medical treatment) | × |
| Family history of SCD | √ (Family history of SCD in ≥1 first-degree relatives <40 years or SCD in a first-degree relative with confirmed HCM at any age) | √ (Family history of SCD in ≥1 first-degree relatives or other close relatives with ≤50 years)(class of recommendation 2a) |
| Unexplained syncope | √ | √ (class of recommendation 2a) |
| NSVT | √ (at least 3 consecutive ventricular beats at ≥120 bpm and <30 s duration) | √ (≥3 repetitive episodes of at least 3 ventricular complexes or ≥1 prolonged episode of NSVT - ≥10 complexes- with at ≥130 bpm. (Class of recommendation 2b.) |
| LV ejection fraction <50% | × | √ (class of recommendation 2a) |
| LV apical aneurism | × | √ (class of recommendation 2a) |
| LGE | × | √ (≥15% of LV mass)(class of recommendation 2b) |
Abbreviations: ACC/AHA: American College of Cardiology/American Heart Association LGE: late gadolinium enhancement; LV: left ventricle; NSVT: nonsustained ventricular tachycardia; SCD: sudden cardiac death. Class of recommendation 2a (moderate): benefit>>risk; Class of recommendation 2b (weak): benefit≥risk.
Regarding ACC/AHA/strategy, the presence of any of the aforementioned risk factors may be an indication for prophylactic ICD implantation, with two different strengths of recommendations (2a and 2b), level of evidence B-NR: moderate-quality evidence from ≥1 well-designed, well executed nonrandomized studies, observational studies or registry studies.
Several independent validation studies followed. Maron et al.32 reported, based on the evaluation of this algorithm in a cohort of 1629 patients, a high specificity for patients with low risk of SCD but a low sensitivity for patients with SCD events, concluding that the European strategy minimizes the overtreatment of HCM patients with ICD, however, leaves a significant percentage of patients at high-risk of SCD unprotected. Conversely, O’Mahony et al.33 performed a meta-analysis involving 7291 patients, confirming that the HCM Risk-SCD model's ability to predict SCD events in low risk (predicted<4%) and high-risk groups (predicted≥6%), with a pooled prevalence of SCD endpoints of 1.01% and 8.4%, respectively. Although this meta-analysis suggested an overestimation of risk in the intermediate group, which presented a pooled prevalence of SCD endpoints of 2.43%, at the end, more than two thirds of SCD occurred in the intermediate and high-risk groups, and, accordingly, the pragmatic approach is to continue to target these patients for primary prevention ICD.
Recently, the enhanced American strategy for prevention of SCD in HCM patients34 and American College of Cardiology (ACC)/American Heart Association (AHA) guidelines29 were published, modifying the American algorithm to redefine the importance of the following markers, now considered to be of high risk: The presence of LV apical aneurysms, extensive LGE in CMR (usually ≥15% of LV mass) and LV systolic dysfunction (defined as LV ejection fraction <50%). Together these risk markers were associated with more than 25% of appropriate ICD therapies in this study. The presence of any of these risk factors or the former ones (with the exception of the ABPRE, which was excluded from the new algorithm) may be an indication for ICD implantation, with different levels of strength. In the group of patients >60 years of age, known to be associated with a low likelihood of SCD,35 the decision to implant an ICD for primary prevention should be individualized.
In a conceptual analysis of the two strategies, one of the main strengths of the European score is the inclusion of continuous variables as they are naturally, since it is unlikely that a septum with 29 mm portends a significant different SCD risk when compared to a 30 mm septum. In addition, the HCM Risk-SCD model incorporates age, which is known to be associated with ventricular arrhythmias,10 and provides an individualized and objective SCD risk prediction. On the other hand, the enhanced American strategy includes new robust risk factors such as systolic disfunction, presence of apical aneurysm and LGE amount, which seem to increase substantially the accuracy of this algorithm (Table 2).
In the absence of a perfect stratification system, both strategies should be applied and discussed and, importantly, the patient's preference should always be considered in a shared decision making process. In controversial cases, other less consensual emerging risk factors may be important and could work in the future as referees, although their role in decision making has not been established (Table 3).
Nonconventional risk markers in hypertrophic cardiomyopathy.
| Nonconventional risk markers | Emerging evidence and reference |
|---|---|
| Gene mutations | • During long-term follow-up, sarcomere positive status was an independent risk factor for SCD/aborted SCD.62• SCD risk was significantly higher in patients with sarcomeric mutations; ventricular arrhythmias were more frequent in HCM patients with MYH7 mutations (vs MYBPC 3 or mutation negative patients).20• Patients with pathogenic or likely pathogenic mutations had a higher risk of malignant ventricular arrhythmias, when compared with patients without any identified mutation.10 |
| Electrocardiographic patterns | • Fragmented QRS was a strong independent predictor of ventricular arrhythmic events and major arrhythmic events.63• Fragmented QRS in ≥3 territories and QTc duration are associated with ventricular tachyarrhythmias/SCD in HCM patients, independently of and incremental to conventional SCD risk factors.64• Pseudo-STEMI pattern, QRS duration ≥120ms and low QRS voltages were independent predictors of SCD or surrogates.65 |
| NSVT duration/rate | • In HCM patients with ICD, faster rate (>200 beats per minutes), longer (>7 beats), and repetitive runs of NSVT were more highly predictive of ICD-treated VT/VF than other NSVT.66 |
| GLS | • Abnormal LV-GLS associates with adverse composite outcomes and ventricular arrhythmias.67 |
| Mechanical dispersion | • Mechanical dispersion was a strong independent predictor of ventricular arrhythmias and related to the extent of fibrosis.68 |
| Microvascular ischemia | • Ischemia-related stress echo criteria were significantly related to outcome (death for all causes, heart transplantations, sustained ventricular tachycardia, acute heart failure and atrial fibrillation).69 |
| LGE presence/quantification | • LGE presence was associated with increase in SCD risk; after adjusting for baseline characteristics, extent of LGE was strongly associated with SCD risk.70• Extent of LGE was associated with an increased risk of SCD events. LGE≥15% was associated with increase in SCD event risk in patients otherwise considered to be at lower risk.71 |
| CMR Postcontrast T1 time | • Post contrast ventricular T1 relaxation time was associated with ventricular arrhythmias.72 |
| Fractional anisotropy | • Low diastolic FA in HCM was associated with ventricular arrhythmias.28 |
| Cardiopulmonary exercise test | • VE/VCO2 slope might improve SCD risk stratification, particularly in those HCM categories classified at low-intermediate SCD risk according to contemporary guidelines.73 |
| Biomarkers | • MMP-9 is a useful biomarker for fibrosis and cardiac events in female HCM patients, whereas MMP-3 is associated with a higher event rate independent from fibrosis and sex.74• Both natriuretic peptides and troponins predict clinical risk independently of established risk factors, and their prognostic power is additive.75• Copeptin and NT-proBNP levels were significantly higher in patients with obstructive HCM, and higher levels were associated with worse outcome.76 |
Abbreviations: CMR: cardiac magnetic resonance; FA: fractional anisotropy; GLS: global longitudinal strain; HCM: hypertrophic cardiomyopathy; LGE: late gadolinium enhancement; LV: left ventricle; MMP-9: matrix metalloproteinase 9; MMP-3: matrix metalloproteinase 3; NT-proBNP: N terminal – pro Brain natriuretic peptide; NSVT: non-sustained ventricular tachycardia; SCD: sudden cardiac death; STEMI: ST segment elevation myocardial infarction; VE/VCO2 slope: ventilation versus carbon dioxide relation during exercise.
Hypertrophic cardiomyopathy research has been strongly focused on risk stratification and prevention of SCD. Consequently, important progress has been achieved in this area and the coexistence of appropriate therapies has drastically reduced mortality from SCD in HCM. HF is now emerging as an increasingly prominent management issue. In fact, HF is an important cause of HCM-related morbidity and mortality, and is a frequent pattern of disease progression.
Melacini et al.36 studied a cohort of 293 HCM patients, assessed between 1980 and 2001 at a HCM referral center, and during a median follow-up of 6 years they detected the presence of severe progressive HF (New York Heart Association (NYHA) class III or IV) in 50 of these patients (17%). Three different HF profiles were defined, based on the predominant pathophysiological component:
- 1)
End-stage systolic dysfunction, defined as LV ejection fraction <50% (n=15, 30% of HF). In this group, LV was more dilated than in the others and had extensive areas of replacement fibrosis, often transmural, probably a consequence of microvascular ischemia, whereas in the other groups, there was either no fibrosis or this was only focal. Natural history of this subgroup of patients was addressed by Harris et al.,37 reporting poor outcomes in 66% of these patients, who died from progressive HF, had sudden death events or underwent heart transplant in a mean follow-up of 3.3 years. Time from HCM symptom onset to systolic disfunction was 10 to 14 years, but time from systolic disfunction to death or heart transplant was 2.7 years. Risk factors for systolic disfunction were younger age at HCM diagnosis, more severe symptoms at presentation, larger LV cavity, less frequent LV outflow tract obstruction (LVOTO), more frequent family history of both HCM (70%) and end-stage HCM (20%).
- 2)
Obstructive (O)-HCM (n=11, 22% confidence interval). In this group, patients were older at the time of HCM diagnosis and LV was more hypertrophied when compared to other HF groups.
- 3)
Nonobstructive (NO)-HCM with preserved systolic function (n=24, 48% of HF). Diastolic dysfunction was common in this group. These patients showed the most accelerated progression to advanced HF, four to six years after symptoms onset, and to adverse outcome defined as death or transplant or last follow-up, another four to six years. In histopathological analysis, LV was hypertrophied and nondilated, with no significant fibrosis. NO-HCM with preserved systolic function and advanced HF patients were also assessed by Rowing et al.,38 in a study including 46 NO-HCM pts who were listed for heart transplant. Twenty of these patients had LVEF≥50%. Similar to Melacini's report, these patients had hypertrophied and nondilated LV, most of them with little or no fibrosis in CMR, and evidence of diastolic dysfunction according to echocardiographic criteria. Family history of HCM was present in 14 of the 20 patients. Interestingly, in five cases (25%), first-degree relatives developed “burn-out” HCM, suggesting a familiar HCM clustering of higher risk HCM, but not equivalent phenotypes.
Concerning HF related to O-HCM, specific treatment options are available, such as surgical myectomy and alcohol septal ablation: in experienced centers, both have demonstrated being very effective and have improved HF morbidity and mortality in O-HCM patients. However, there is a gap in specific therapy for NO-HCM-related HF, perhaps because its molecular basis is still not well understood. The just published AHA/ACC guidelines29 recommend treating HF symptoms in patients with NO-HCM similarly to other patients with HF. However, the literature demonstrates that this group continues to present a worse prognosis when compared to other groups of HCM patients. Intensive research is needed to better treat these patients.
Atrial fibrillationAtrial fibrillation is a frequent complication of HCM and has historically been associated with increased morbidity and mortality in these patients.39,40 However, more recent studies41 suggest, under the current treatment strategy, that AF is not an important contributor to HF or a cause of SCD. In fact, AF is rarely the primary cause of death in HCM and is almost virtually limited to the context of embolic stroke, which supports a low threshold for starting anticoagulant therapy. Rowing et al.41 report no difference in the outcomes of patients with HCM and AF vs. HCM without AF, comparing age and sex-adjusted cohorts, regarding SCD, global or HCM-related mortality or NYHA class progression. Although each episode of paroxysmal AF can manifest itself with great clinical exuberance, the decrease in a patient's quality of life is transient and treatable, and arrhythmia will not inevitably be progressive. Only 25% of patients progress to permanent AF.
Atrial fibrillation treatment in patients with HCM also has some specificities: a take-home message is, in the absence of contraindications, there must be a low threshold to start anticoagulation, usually after the first episode of AF. Guttmann et al.42 developed a prediction model in HCM for thromboembolic events (TE) at five years based on the following clinical variables: age, AF, previous TE, NYHA class, left atrium (LA) diameter, vascular disease and maximum LV wall thickness. Although it has not been demonstrated that patients with HCM in sinus rhythm with a high estimated risk of TE should undergo anticoagulation prior to the first AF episode, the recommendation of frequent ECG monitoring in an outpatient setting in patients with dilated LA is supported. In fact, the authors demonstrate a linear relationship between the diameter of the LA and TE risk until 45-50 mm. Above that cut-off, TE risk increases exponentially. In contrast, the CHA2DS2-VASc score has a very low predictive acuity in this population and should not be used in HCM.
Despite the scarcity of randomized trials on the use of new oral anticoagulants in HCM, data from large observational studies43–45 suggest a non-inferiority of these new agents in relation to warfarin, associated with a greater comfort of administration. Indeed, current AHA/ACC guidelines29 recommend direct-acting oral anticoagulants as a first line option in these patients.
Drug selection for rhythm maintenance in patients with HCM is based on extrapolation from studies on AF in the general population. Most recent HCM recommendations29 consider reasonable therapy with amiodarone, dofetilide or sotalol. However, current evidence in AF-HCM patients suggests rhythm maintenance pharmacological therapy has limited efficacy, especially over time.46
Similarly, AF catheter ablation does not appear to be as effective as in non-HCM AF patients.47 Even so, current evidence suggests an improvement in the outcome of patients with HCM and AF undergoing ablation vs. those controlled with pharmacological therapy.48 A meta-analysis including five comparative studies of AF ablation success in HCM vs. non-HCM patients49 reported that patients with HCM and AF had a two-fold increase of risk of recurrence of arrhythmia compared to non-HCM patients. However, the outcome in patients with HCM, with less dilated atria and paroxysmal AF appeared to approximate that of the general population. Since the risk of procedure-related adverse events was low, the authors conclude that catheter ablation may be effective in patients with AF and HCM, particularly in patients with small atria.
New pharmacological agentsTo date, recommended pharmacological therapy in patients with symptomatic O-HCM has been nonspecific and had limited benefits on symptom relief. In patients with moderate to severe symptoms, in spite of maximally tolerated drug therapy, invasive treatment to reduce LVOTO should be considered, surgical septal myectomy or alcohol septal ablation. Both strategies have a very high success rate in experienced centers, although neither is risk-free and some patients require multiple interventions, especially those who undergo alcohol septal ablation.
Mavacamten in O-HCMThe pathophysiological mechanism involved in the development of LVOTO includes an hyperdynamic ejection and an exacerbated inotropism that, in HCM patients, results from an exaggerated interaction between actin and myosin filaments. This process results in hypercontractility, incomplete relaxation and increased waste of energy, which, ultimately, through several still unknown pathways, leads to myocardial dysfunction and fibrosis.
Mavacamten is a specific allosteric inhibitor of cardiac myosin ATPase, which has been shown in vitro to be able to restore the proportion of myosin in a super relaxed state, reducing excess interaction between the myosin and actin filaments and normalizing ATP consumption.50 Pre-clinical studies in HCM animal models demonstrated that the administration of mavacamten reduced myocardial contractility, eliminated mitral valve systolic anterior movement and decreased LVOT gradient and, if administered before the implementation of the hypertrophic phenotype, attenuated the development of left ventricular hypertrophy, myocardial disarray and fibrosis.51
The recently published EXPLORER-HCM multicenter phase 3 study aimed to assess the efficacy and safety of mavacamten in the treatment of symptomatic patients with O-HCM (NYHA class II or III).52 This clinical trial, which involved 251 patients, 123 receiving mavacamten and 128 placebo, lasted 30 weeks and was completed by 97% of patients. Mavacamten lead to a significant decrease in LVOT gradients after exercise. Almost 75% of patients showed a reduction to values <50 mmHg and 56% demonstrated complete disappearance of the obstruction. Mavacamten also demonstrated improvement in NYHA functional class, in peak VO2, in symptoms and quality of life assessed scores, and significant reductions in N terminal-pro nrain natriuretic peptide (NT-proBNP) and troponin I levels. Mavacamten was well tolerated, with a safety profile comparable to placebo. It is, therefore, a promising drug in O-HCM patients.
Mavacamten in NO-HCMBased on in vitro tests that demonstrated the ability of mavacamten to reduce the interaction between myosin and actin filaments, it is possible that, through an improvement in diastolic function and an optimization of energy use, this new drug may be beneficial in a NO-HCM patient population. The MAVERICK-HCM study53 was a multicenter phase 2 study designed to evaluate the safety and tolerability of mavacamten in symptomatic patients (NYHA class II or III) with NO-HCM. Mavacamten was well tolerated by most patients: serious adverse events were detected in 10% of the group undergoing treatment vs. 21% in the placebo group, and in all cases, patients recovered without sequelae. In five out of 40 patients receiving mavacamten, EF decreased to values ≤45%, which was reversible after drug suspension. In addition, therapy was associated with a significant decrease in NT-proBNP and cardiac troponin-I which suggests an improvement in myocardial stress, paving the way for phase 3 studies to seek clinical benefit in this patient population.
ExerciseUntil very recently, an HCM diagnosis imposed an almost total restriction on intensive sports, based mainly on a study of causes of sudden death in young American athletes, which suggested that exercise increased the risk of SCD in patients with HCM.54 However, recent longitudinal studies seem to indicate that the risk of SCD during exercise is lower than previously considered. Pellicia et al.55 in a study with nine years of follow-up, found no difference in the incidence of symptoms or major events in athletes with HCM who stopped physical exercise (n=20) vs. athletes with HCM who continued to practice competitive sport (n=15). On the other hand, in a cross-sectional study involving 121 patients with HCM, the practice of vigorous physical exercise was similar in groups with or without ventricular arrhythmias.56
Current recommendations57 adopt a more liberal approach to sports participation in some patients with HCM, especially those who intend to maintain non-competitive physical activity. In fact, the most recent AHA/ACC guidelines29 comment on recreational exercise as having benefits on general health in HCM patients, similar to that of the general population. A rigorous evaluation must be carried out by experts, assessing HCM severity but also excluding other comorbidities such as hypertension or coronary heart disease. Recommendations57 advise against the practice of high intensity exercise for individuals who present any marker of increased risk, such as 1. History of SCD or unexplained syncope; 2. Estimated five-year risk of SCD≥4% using the HCM Risk-SCD model; 3. LVOTO at rest >30 mmHg; 4. ABPRE; 5. Exercise-induced arrhythmias. For these patients, the practice of recreational exercise of low to moderate intensity may be considered. In HCM patients without any of the aforementioned risk factors, practice of competitive high intensity physical exercise may be considered, with the exception of cases in which the occurrence of syncope may result in injury or death.
Also relevant in this area is another study underway, which is attempting to characterize the impact of physical exercise and lifestyle on the well-being of HCM patients (LIVE-HCM). Until accurate answers are obtained, the discussion of the clinical context with the patient and shared decision making is accepted as the best strategy for applying restrictions to exercise in this population.
ConclusionHypertrophic cardiomyopathy is a complex and heterogeneous disorder, still presenting with significant morbidity and mortality. Data from recent registries suggest the existence of two HCM subgroups (sarcomere positive vs. nonfamilial) with different etiologies, histopathologies, clinical presentations and prognosis. Future research should clarify the importance of using this classification to treat better these subgroups of patients who may require specific management strategies.
Recent advances in SCD risk stratification and prevention have drastically reduced SCD events, making HF emerge as a significant management issue in HCM population. Regarding O-HCM HF therapy, invasive LVOTO reduction therapies are available, with proved effectiveness, and promising non-invasive therapies are being investigated in clinical trials. Currently, there is still a gap in specific therapy for NO-HCM, which continues to be a less well understood entity.
Future research should focus on better understanding HCM disease mechanisms to treat specifically HCM, a paradigm of translational medicine.
