



Iron interactions with the cardiovascular system were proposed about half a century ago, yet a clear-cut understanding of this micronutrient and its intricacies with acute and chronic events is still lacking. In chronic heart failure, patients with decreased iron stores appear to benefit from intravenous administration of metallic formulations, whereas acute diseases (e.g., myocardial infarction, stroke) are barely studied in randomized controlled trials in humans. However, proof-of-concept studies have indicated that the dual redox characteristics of iron could be involved in atherosclerosis, necrosis, and ferroptosis. To this end, we sought to review the currently available body of literature pertaining to these temporal profiles of heart diseases, as well as the pathophysiologic mechanism by which iron enacts, underlining key points related to treatment options.
As interações do ferro com o sistema cardiovascular foram propostas há cerca de meio século; falta ainda, no entanto, uma perceção clara sobre esse micronutriente e sobre as suas complexidades nos eventos agudos e crónicos. Na insuficiência cardíaca crónica os doentes com diminuição das reservas de ferro parecem beneficiar da administração intravenosa de formulações metálicas, enquanto as patologias agudas (por exemplo, enfarte do miocárdio, AVC) são pouco estudadas em ensaios controlados aleatorizados em humanos. No entanto, estudos de prova de conceito indicaram que a dupla característica redox do ferro poderia estar implicada na inaterosclerose, necrose e ferroptose. Para tal, procurámos rever o atual conjunto de literatura disponível sobre estes perfis temporais de doenças cardíacas, bem como o mecanismo fisiopatológico pelo qual o ferro exerce os seus efeitos, sublinhando pontos-chave relacionados com as opções de tratamento.
Cardiovascular diseases are one of the leading causes of death in the modern era of medicine; heart failure (HF) is one of the major stepways common to a plethora of such diseases. In 2016, a third of deaths was attributable to cardiovascular diseases, with the greatest culprits being ischemic heart disease and stroke.1 Whereas disability-adjusted life years owing to cardiovascular disease have risen to the first place compared to 2000.2 As the increase in longevity of the global population continues, new challenges and therapeutical targets are being pursued in cardiovascular diseases, particularly in the context of HF and its ancillary diseases.
Normal iron levels are paramount to a healthy individual, as their metabolism is involved in erythropoietic cell function with rapid turnover rates, as well as in the normal functioning of cellular enzymes and organelles. As such, iron replenishment is advised for HF patients and those with reduced ejection fraction with or without anemia, but with established decreased blood iron levels; although given the potential for increased reactive oxygen species (ROS) generation3 and inflammation, concerns regarding effects of such ambivalent micronutrient have merit.4 Roughly a quarter of patients who suffer an acute cardiovascular event [such as myocardial infarction (MI), episodes of ischemia/reperfusion (I/R), stroke] progress to overt HF.5 While the administration of iron appears beneficial in the chronic milieu, its effects in atherothrombotic occurrences seem less encouraging, thereby this review brings into focus the potential implications of therapeutical iron approaches and cardiovascular diseases in an acute and chronic setting.
Effects of iron in cardiovascular homeostasisIron is an indispensable element for biological life and is used at the molecular level for mitochondrial ATP production, DNA synthesis, and oxygen ferrying in the erythrocytes.6 Its two valences enable redox activity, with the Fenton reaction inducing the generation of ROS along with deleterious effects on the cell's metabolism. The sites of action for iron and iron moieties (heme, iron-sulfide) are diverse, ranging from the cell cytoplasm,7 membrane,8 to the mitochondria.9 The physiologic pathway of iron begins in the gut, with ingress at the apical border of the enterocyte. Following hepcidin-dependent absorption into the bloodstream, iron is delivered to cells via membrane transferrin receptors and is further regulated within the cell by specific molecular mechanisms.10 The systemic control of iron homeostasis is regulated in an endocrine manner through hepcidin release from the liver, which limits the amount of iron absorbed if iron deposits are adequate or in case of inflammation/infection.11 In turn, hepcidin is controlled by oxygen levels, cell proliferation status, erythropoiesis requirements. Intracellularly, iron response proteins influence the fate of iron handling.12
In the setting of the cardiovascular system, the effects of iron are increasingly noted both in the case of deficiency and excess, and13,14 are involved in pathophysiologic events. The putative involvement of the mechanism of necrosis and organ failure during ischemia/reperfusion (I/R) has been under scientific scrutiny. Whereas iron supplementation was found to be at least symptomatically beneficial for established HF,15 increased iron deposition was found to be partly responsible for the development of I/R injury.16
Iron deficiency is postulated to be one of the causes of deficient energy production within the cardiomyocytes, causing undue imbalance between the required parameters for adequate perfusion and output capacity of the heart, resulting in HF.17 In the chronic setting of inflammation, hepcidin is increased (causing decreased iron availability), as it possesses anti-microbial and anti-inflammatory effects.18 It appears that the continuous release of hepcidin during prolonged inflammation leads to a loss of homeostasis and its replacement with a pathological frame, in which this acute-phase protein causes starvation of the metallic moiety.19 Therefore, a large proportion of patients are either iron-deficient or anemic – albeit independently of iron status (i.e., red cell indices do not show features characteristic of iron deprivation).20
However, iron presence in the metabolic processes that take place in an acute setting appears to have deleterious effects,21 particularly through ROS generation and ferroptosis-induced cell death. Additionally, the presence of hepcidin during acute processes seems to be of physiologic benefit, protecting the injured tissue from excessive harm caused by iron.22 A growing body of data suggests that iron might be involved both directly in the actual process of ischemia4 and at an etiological level, in the development of atherosclerosis.23,24 While ischemic heart disease imposes the heaviest burden in HF, its connection with iron metabolism has been rarely assessed. A recent study concerning blood iron levels and their correlation with adverse cardiovascular events indicated that patients with ischemic HF are at increased risk if they are situated in the higher echelons of serum iron.25 Conversely, a cohort study (206 patients and 436 controls)26 presented findings that link low serum to a short-term risk of developing cardiovascular disease. These findings must be interpreted with caution considering that systemic iron levels (i.e., serum iron) do not correlate well with cardiac iron levels.27
Soluble transferrin receptor, a novel endogenous marker which could reflect intracellular iron requirements and predict cardiovascular disease progression28 might be a more useful parameter to quantify iron-heart interaction. The soluble form appears in individuals’ sera as a marker of decreased cell iron availability. Additionally, the configuration in which iron circulates could stem its two-sided profile, as the increased presence of its non-transferrin-bound form induces atherosclerosis and inflammation,24 while chaperoned iron could be of beneficial use.29,30
FerroptosisAccidental cell death is regarded as unavoidable and is defined by extreme variations in the physical and chemical parameters (burns, trauma), while regulated cell death is either physiological (programmed cell death) or induced by stressors. These in turn activate molecular mechanisms that bring about the cell's demise (as opposed to the lack of mechanisms in accidental cell death).
Ferroptosis (ferro, “iron”; ptosis, “falling”) is iron-modulated necrosis, consisting in the accumulation of lipid peroxidation products and ROS,31 likely caused by either increased iron bioavailability or by excessive ROS, as shown by inhibition of ferroptosis by iron chelation and ferrostatin.9,31 At the molecular level, blocking of the system XC− antiporter (cystine and glutamate) is principally involved in the development of lipid peroxidation, with its respective antagonistic agents (erastin, glutamate, sulfasalazine) inducing ferroptosis.31 However, iron species involvement is indirect (yet essential), purportedly via Fenton reaction-induced ROS. It is relevant to mention that iron per se is also directly involved in cell death (without the participation of ROS/ferroptosis), with nascent therapeutic implications in a cardiovascular setting.32 Furthermore, the acyl-CoA synthetase long-chain family member 4 (ACSL4) is pivotally implicated in providing the necessary fatty acids required for ferroptosis.33 Interestingly, the fatty acids that are required for ferroptosis are polyunsaturated fatty acids (PUFAs), a staple of lipids recommended by guidelines34 to reduce cardiovascular risk, raising the possibility of a double-edged sword effect. Being mediated through toxic lipid peroxidation, coupled with research data pointing at increased iron involvement in atherosclerosis,17,24 ferroptosis is most likely implicated in the development of atherosclerosis, with inhibitors of ferroptosis providing protection in a mouse model of dyslipidemia-induced atherosclerosis.35
Fortuitously, ferroptosis appears to be already clinically tenable to pharmacologic modulation. Inhibitors of ACSL4 (and of ferroptosis, concurrently) are also an antidiabetic class of drugs – thiazolidinediones, whereas vitamin E and deferoxamine (an iron chelator) exhibit their protective effects within the mitochondria, at the site of lipid peroxidation.36
Acute cardiovascular eventsStroke, myocardial infarction, vascular dissections and thromboembolisms are acute stressor events on the cardiovascular system that pose a two-fold challenge: the abrupt cessation of blood flow brings about the inability of host capacity for protection during the event, while the loss of function has a long-lasting and profound effect on organ function. This means that the injury present in acute events is arguably more difficult to treat, reverse, and institute preventive measures for. In recent years, studies in the setting of ischemia have shed light on the shifts in the mitochondrial metabolism during oxygen deprivation and overflow. This will be discussed briefly in the subsequent paragraphs.
Ischemia/reperfusionThe advent of reperfusion strategies during acute coronary occlusion has led to a reduction in adverse outcomes; however, this abrupt inflow of oxygen-ridden blood through the ischemic tissue leads to further cell damage and necrosis via additional mechanisms, prompting the need for further description of the pathophysiological processes that are involved, as well as potential targets for clinical therapy.
It appears that ischemic injury is brought into sequence by disrupting the yield of aerobic metabolism via dysfunctional ATP-linked enzymes that in turn increase intracellular acidity and calcium levels, mitochondrial permeability transition pore opening and mitochondrial iron uptake, myofibril hypercontracture. The culmination is membrane rupture via a plethora of non-programmed cell death mechanisms,37 whereas reperfusion expounds its deleterious effects by means of oxygen conversion to ROS (via NADPH oxidases family), calcium overload, aided by dysfunctional enzymes responsible for modulation of oxidative stress (such as GPX4, reduced coenzyme Q10)38 (Figure 1).
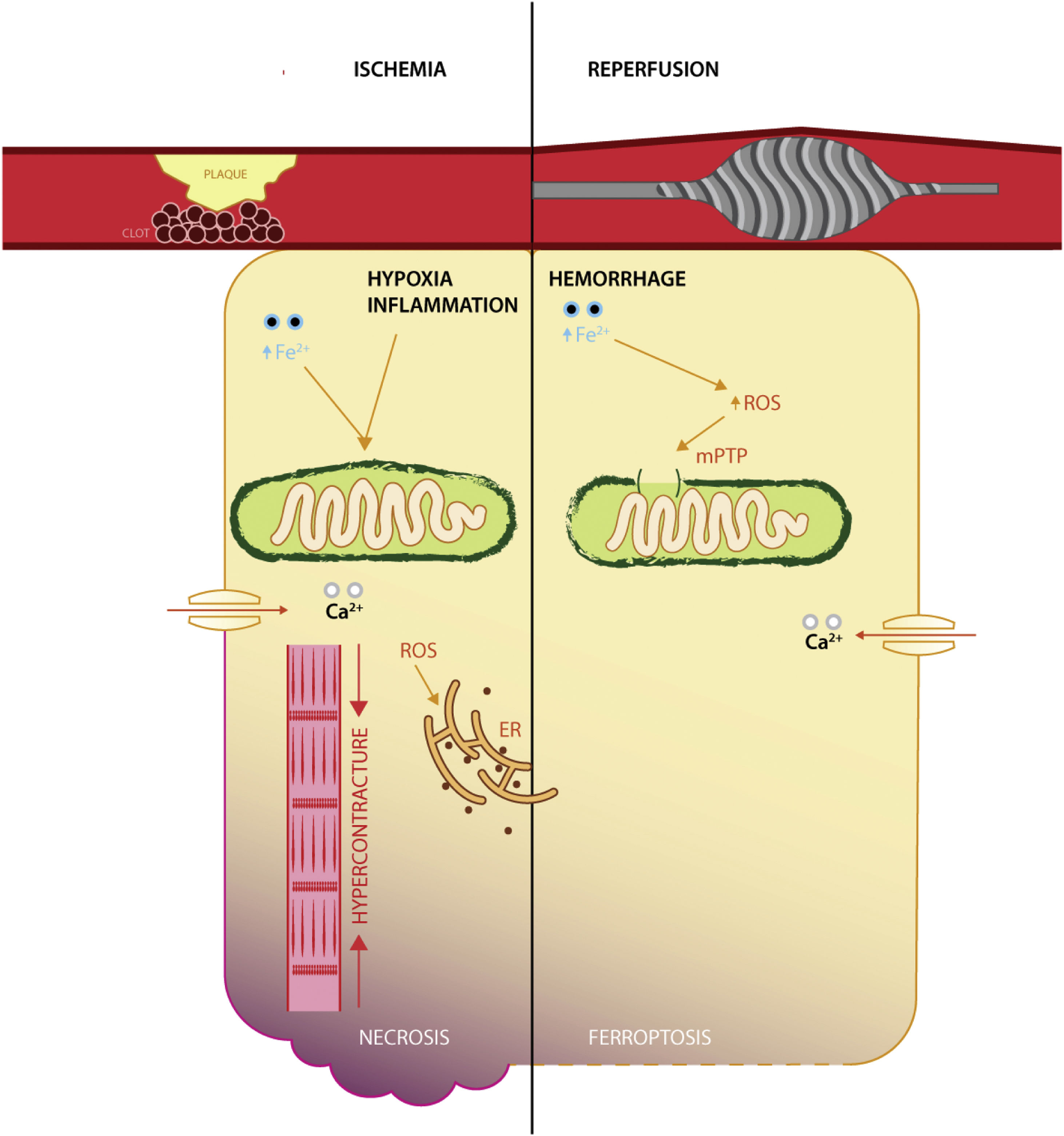
Interestingly, a recent study in a rat model of I/R has shown that reperfusion, rather than ischemia, induces ferroptosis, with the degree of necrosis being amenable by iron chelation.4 It appears that following acute coronary syndrome, both ischemia and reperfusion induce iron deposition in the infarcted areas, with increased affinity for reperfused, oxygen-rich tissues.39 While the relative success of reperfusion (as appraised by electrocardiographic changes) is abated in patients with sera rich in iron transport proteins and inflammation.40 Furthermore, the relative protection conferred by iron deficiency, on short-term survival, at the time of an ST-elevation myocardial infarction, reveals the enhanced interaction in the reperfusion phase.41 This pinpoints the potential clinical use of iron therapeutical manipulation in the catheter laboratory.42
Diabetes and acute coronary syndromes are well established comorbidities frequently found together in a substantial proportion among cardiovascular patients.43 Iron deposition occurs both in the pancreas and in the heart,44 relating the two conditions in an iron-dependent paradigm. ROS production in the setting of both end-glycation products and reperfusion injury could plausibly induce in greater magnitude cell death via ferroptosis. A specific example is that of an animal model of diabetes and myocardial I/R which showed that at least part of the ischemic offense is mediated through increased endoplasmic reticulum stress, rather than solely the canonical mitochondrial site.7
The effect of iron on myocardial I/R seems to be mediated by increased inflammation, edema, and deleterious remodeling of the heart chambers, especially in areas affected by hemorrhage, as opposed to microvascular obstruction,21 consistent with the relative lack of involvement in the ischemic phase. Reperfusion injury is also greater if the capacity to scavenge hemoglobin-bound iron is diminished, by decreased function of haptoglobin phenotyping.45 While there certainly is a lack of human translational purviews, at least one clinical study has presented evidence of increased all-cause mortality in patients with elevated circulating iron at the time of an acute coronary syndrome.46 Thus, the reperfusion phase is a potentially viable candidate for therapeutical modulation, aligning temporally with the use of cardioprotective agents that are usually administered chronically post-acute event, rather than before or during the ischemic phase (which seldom happens).
Mitochondrial involvementReactive oxygen species in the mitochondria induce cell death3 by disrupting the electron transport chain and decreased mitochondrial membrane potential that yields a precarious production of ATP, which is unable to sustain cell homeostasis. Iron is meant to enact its biological effects mainly within mitochondria, where it interacts with cell respiration, ROS production, and iron-sulfur clusters.47 The mitochondrial apparatus is one of the main sources of ROS, as a residual product during oxidative phosphorylation, which could explain the increase of oxidative stress and inflammation during ischemia/reperfusion.48 Additionally, the localization of important antioxidant enzymes, such as GPX4 (see above) is also confined to the mitochondria, linking ferroptosis with acute cardiovascular diseases.
The pivotal event in the mitochondria during I/R injury is the modulation of the mitochondrial permeability transition pore, which remains closed during ischemia, but opens during reperfusion, causing a reduction in mitochondrial membrane potential and of oxidative phosphorylation.49 Furthermore, the decrease in oxidative phosphorylation leads to increased intracellular acidity and consequent hypercontracture via calcium excess.50
Ferroptosis contributes to acute ischemia by way of lipid peroxidation. In this regard, aldehydes, formed by lipid peroxidation, disturb the normal function of redox modulatory systems within the mitochondria (especially within its membranes), with the endogenous aldehyde dehydrogenase 2 decreasing ROS generation in animal models of cardiac arrest.51
Mitochondrial ferritin is regarded as an antioxidant molecule, with a similar composition to H (heavy) ferritin found within the cytoplasm, although it appears that it is not modulated by iron levels.30 In a particularly relevant oxidative damage experiment, acute myocardial oxidative stress in mice lacking myocyte mitochondrial ferritin further increases levels of labile iron, with deleterious effects regarding cell survival, energy production, and lipid peroxidation.29
Clinically, studies regarding mitochondrial involvement are sparse; still, in regard to patients with chronic HF and reduced ejection fraction, iron administration induces a better energetic profile in skeletal muscles, alleviating exercise capacity and functional class.52
Chronic cardiovascular diseaseChronic heart failure (CHF) is a persistent condition in which the chief complaints of the affected individual correspond to a structural or functional abnormality of the heart which results in decreased tissue perfusion or increased ventricular wall stress.15 Patients have a plethora of comorbidities, mainly anemia and/or iron deficiency (ID),17 which are related to worse outcomes. Iron dysregulation is frequently encountered in this population of patients, the etiology being multifactorial. Definitions of ID in chronic inflammatory diseases are a point of contention, whereas the treatment options in the form of intravenous iron preparations show promising results.
Etiology of iron deficiencyDecreased absorption within the gutAlthough traditionally thought to be a determinant of ID,53 with congestion being prevalent in a low output state and poor results in patients receiving oral iron,54 current studies suggest a lesser correlation between the two.55 A plausible explanation resides in the heart's specific iron handling pathway, by way of cardiac hepcidin.56 Additionally, the capacity to alleviate anemia in CHF patients seems similar between oral and intravenous formulations,57 further supporting the paracrine regulatory mechanism.
Nutritional deficitMicroelement intake is dysregulated not only regarding iron, but also other essential metals.58 In line with this, lower protein intake, as well as low serum albumin, correlate well with the presence of ID.59 Malnutrition could be related to decreased appetite, ascites, and gastrointestinal symptoms.60
InflammationChronic inflammatory states, such as diabetes, chronic obstructive pulmonary disease, and inflammatory bowel syndrome share the TNF-alpha and interleukin-mediated inflammation.61,62 The interplay with hepcidin impedes he systemic bioavailability of iron, as well as increased activation of the RAAS, causing a vicious cycle.14 However, disappointing results with anti-inflammatory modulation in CHF as well as an unclear causal relationship in HF led to equivocal conclusions regarding inflammation as etiology of ID.63 Certainly, a degree of involvement is bound to exist, as well-constructed studies show a correlation between established markers of inflammation and HF patients, with yet undiscovered pathophysiologic mechanisms.64
Definition of iron deficiencyThe current guideline-based approach suggests that absolute ID is defined by serum ferritin <100 ng/mL, while functional deficiency was allotted at ferritin levels between 100 and 299 ng/mL with a transferrin saturation of less than 20%.15 Debate has arisen regarding the definition, with a swath of studies that have correlated more precisely with cardiovascular hard endpoints (e.g., mortality). Increased mortality and risk for hospitalization were seen with transferrin saturation <20%, but not with isolated ferritin,27,65 a pattern also observed in patients undergoing hemodialysis,66 indicating a potentially harmful effect of injudicious iron treatment. Furthermore, serum indices of iron are poorly corelated with myocardial iron content and availability,55,67 deluding clinicians in tailoring appropriate therapies for appropriate patients. Additional parameters for ID diagnosis such as soluble transferrin receptor, hepcidin, erythroferrone are being considered, albeit studies for efficacy and their clinical implementation are lacking.68
Therapeutical modulation of ironSince the first hypotheses on iron involvement in cardiovascular disease, an ever-increasing number of trials have been conducted to assess the effect of iron modulation (chelation and supplementation), producing very promising results on iron administration (Table 1).
Iron homeostasis and cardiovascular disease in human trials.
| Citation and sponsor | Population (n) | Treatment | Main conclusion | Comments |
|---|---|---|---|---|
| Iron administration | ||||
| 72 Ponikowski et al., 2020Sponsored by Vifor Pharma | Stabilized acute HF, with ID and HFrEF, (1132) | Ferric carboxymaltose, iv. | Reduced hospitalization for HF | Similar adverse effects vs. placebo |
| 77 Nunez et al., 2020;Supported in part by Vifor Pharma | Stable HF with EF <50%, ID and Hb <15 g/dL, (53) | Ferric carboxymaltose, iv. | Increased iron deposition in the heart;Increased quality of life | EF and NT-proBNP did not differ vs. placebo |
| 78 Anker et al., 2009;Supported by Vifor Pharma | CHF, EF <45%, ID and Hb [9.5–13.5] g/dL, (459) | Ferric carboxymaltose, iv. | Improved quality of life, functional capacity and symptoms | Similar adverse effects vs. placebo |
| 79 Ponikowskiet al., 2015;Sponsored by Vifor Pharma | CHF, EF <45%, ID (304) | Ferric carboxymaltose, iv. | Improvement in functional capacity, symptoms and quality of life | Similar adverse effects vs. placebo |
| 80 Spahn, Schoenrath et al., 2019;Sponsored by Vifor Pharma and Swiss Foundation for Anaesthesia Research | Patients undergoing valve- and/or coronary bypass surgery, with ID or anemia, (1006) | Ferric carboxymaltose/erythropoietin/vitamin B12/folic acid | Reduced periprocedural need for transfusions; | Single center RCT;No evaluation of hard end-points; |
| 54 Lewis et al., 2017Sponsored by NHLBI | CHF, EF <40%, with ID and Hb [9–15] g/dL, (225) | Iron polysaccharide, oral | Oral iron did not improve exercise capacity, 6MWT, nor quality of life | Similar adverse effects vs. placebo |
| Iron chelation | ||||
| 81 Yeatts et al., 2013;Sponsored by NIH | Intracerebral hemorrhage, (42) | High dose deferoxamine, iv. | Trial terminated for increased mortality in treatment arm | Dose was consequently decreased in further trials |
| 82 Selim et al., 2019;Sponsored by NIH/NINDS | Acute intracerebral hemorrhage (294) | Deferoxamine mesylate, iv. | Iron chelation did not improve clinical outcome at 90 days post-event | Deferoxamine was safe;This was a phase II trial that did not show merit for a phase III trial |
| 83 Fernandes et al., 2016;Sponsored by Fundacao de Amparo a Pesquisa do Estado de São Paulo | Thalassemia major and myocardial siderosis, (62) | Amlodipine in addition to standard chelation (deferiprone/ deferoxamine) | Calcium channel blockers reduce myocardial iron | Treatment did not affect systemic iron homeostasis |
| 84 Karlsson et al., 2015;Sponsored by Medical Research Council of Southeast Sweden, PledPharma AB | STEMI and primary PCI, (20) | Mangafodipir, iv. | Treatment did not alleviate biomarker burden vs. placebo;Drug was safe to administer | The usual dose for the drug was lower than commonly used;Population size was modest (phase I trial) |
| 85 Chan et al., 2012;Sponsored by National Health and Medical Research Council of Australia | STEMI and primary PCI, (60) | Deferoxamine, iv. | Treatment reduced oxidative stress | The primary end point (infarction size) was not different vs. placebo |
| 86 Paraskevaidis et al., 2005;Sponsored by: not declared | Coronary artery disease and CABG, (45) | Deferoxamine, iv. | Protection against reperfusion injury and lowers ROS | Unclear blinding strategy;Single center, small sample study |
CABG: coronary artery by-pass graft; CHF: chronic heart failure; EF: ejection fraction; HFrEF: heart failure with reduced ejection fraction; ID: iron deficiency; NIH: National Institute of Health; NTproBNP: N terminal pro brain natriuretic peptide; PCI: percutaneous coronary intervention; RCT: randomized controlled trial; ROS: reactive oxygen species; STEMI: ST elevation myocardial infarction.
Oral iron efficacy was surveilled in patients with HF, in whom it was found that oral administration is not adequate to decrease clinical events.54,69 Several trials47,54 indicate that while both formulations increase hematinic parameters, only the parenteral route increases aerobic capacity and energy efficiency. To date, intravenous administration of iron appears the most relevant approach to iron deficiency in CHF. Randomized controlled trials54,70,71 have illustrated the potential benefit in patients with HF and reduced ejection fraction lacking iron, independent of anemia status. Interestingly, patients with established HF but with preserved ejection fraction do not respond in a similar manner to iron administration, a finding supported in part by a different pathophysiologic paradigm and by the lack of trials with this type of patients. Given that the endpoints of the aforementioned trials are lacking in pursuing mortality and major cardiovascular events, clear answers still need to be presented. FAIR-HF2 is specifically designed to observe cardiovascular death and hospitalization for HF, a trecut 2021, poate ar merge aici o actualizare a rezultatelor, while iron supplementation in acute decompensations of established HF patients is scarcely evaluated. AFFIRM-AHF72 is a double-blind, randomized controlled trial regarding iron administration in patients with stabilized acute HF of any etiology and concurrent iron deficiency. The main conclusion of the trial was a 20% reduction in cardiovascular hospitalization compared to placebo, while the primary endpoint of cardiovascular death showed the same relative alleviation, narrowly missing conventional significance (p=0.059). Current knowledge based on human clinical trials appear to favor iron administration with improved secondary outcomes, while generally lacking a consistent reduction in cardiovascular death, inferring that, while iron therapy is useful, the degree of additional protection (on top of already established cornerstone medical treatment with beta-blockers, renin-angiotensin-aldosterone blockers) conferred by this type of therapy is lackluster.
Historically, iron toxicity has been related mainly to its gastrointestinal (for oral preparations) and localized at the site of administration (for intravenous forms). A Cochrane Database review of oral and parenteral preparations cited constipation (RR 1.63), diarrhea (RR 2.17) and nausea/vomiting (RR 1.75) as significant side effects, with a 60% increase in overall risk of side effects (RR 1.60; 95% CI 1.23–2.07; four studies; 1748 participants; p=0.0005).73 Concerning parenteral formulations, especially ferric carboxymaltose, hypophosphatemia is a notable side-effect,74 potentially influencing bone mineralization, cell function and metabolism. The mechanism behind this decrease in phosphate is unclear, but it appears to be downregulated by increased renal excretion via fibroblastic growth factor 23 modulation.75 However, its correlation with the clinical consequences or symptoms appears undiscerning, further complicated by the lack of standardization in methodology and reporting of this parameter.76
Iron chelationThe cellular hypothesis of increased iron, especially in the mitochondrial compartment, raises the issue of treatment-related harm, perchance explaining the current ambiguousness in clinical trials on iron supplementation.14,67 Preclinical models of HF have shown interesting results regarding iron chelation, with improved fibrosis, cardioprotection, and increased mitochondrial energetics.87-89 Arguably the most researched field concerns iron overload cardiomyopathies, where the mainstay therapy is removal of excess iron, either by phlebotomy or iron chelation, where feasible. Primary iron overload occurs in hemochromatosis, as a result of genetic dysregulation of iron handling, and secondary, induced by treatment of hemoglobinopathies, dialysis, or malignancies.90 The majority of patients succumb as a result of myocardial interest and consequent HF. There is a positive correlation between the susceptibility to iron overload and the amount of calcium transporters in the respective tissue, with protective effects of calcium channel blockers.91
The mechanistic process involves ROS production within the cytoplasm and mitochondria, leading to energetic failure.16 Additionally, increased myocardial fibrosis and ferroptosis induce diastolic and systolic dysfunction, and arrhythmias. Approved treatment options vary according to disease; in the case of primary iron overload, blood-letting targeting ferritin between 50 and 100 ng/mL and transferrin saturation below 50% is the current standard,92 whereas in patients where venesection is contraindicated (i.e. anemia), chelation therapy with deferoxamine, deferiprone and deferasirox are safe and effective. Furthermore, L-type calcium channel inhibitors, used both in human and murine studies, provided additional benefits when used alongside mainstay therapy.89 Recently, anthracycline (doxorubicin) induced cardiomyopathy was shown to be caused by increased mitochondrial iron accumulation, with chelation or ferroptosis inhibitors proving to be protective.9
ConclusionsIn the context of basic science, a hypothesis regarding iron interactions with acute vs. chronic conditions can be formulated as a basis for further clinical research (Table 2). Chronic HF is currently approached via iron supplementation, with increasing prospects for beneficial effects, such as improved bioenergetics, anemia correction, reduction in cardiac stress and inflammation, whereas iron chelation appears less well studied, apart from obvious beneficial effects in iron overload cardiomyopathies (thalassemias, hemochromatosis). Poignantly, iron dysmetabolism and its direct correlation with clinical events in human trials is lacking or the results are inconclusive.
Proposed pathophysiology of iron in acute and chronic cardiovascular events.
| Increasing iron | Decreasing iron | |
|---|---|---|
| Acute events | • Increased atherosclerosis23,94* | • Decreased lipid peroxidation95* |
| • Increased I/R injury21* | • Decreased inflammation21* | |
| • Ferroptosis35* | ||
| • Reduced hospitalization in stabilized AHF72 | ||
| Chronic events | • Improving symptoms in HFrEF71 | • Decreasing fibrosis, ROS98* |
| • Improved distance on 6 minute walk test in patients with HFrEF96 | • Mitochondrial protection9* | |
| • Ameliorated cell (including skeletal muscle) energetics in HFrEF97 | ||
| • Decreased hospitalization in HFrEF70 | ||
AHF: acute heart failure; HFrEF: heart failure with reduced ejection fraction; I/R: ischemia/reperfusion; ROS: reactive oxygen species; animal model research is marked with an asterisk (*).
During I/R or MI, decreasing the amount of available iron protects the endothelium and the heart's reperfusion injury, decreasing ROS production and fibrosis,21,24 by inhibition of atherosclerosis and inflammation,93 and down-regulation of oxidative stress (by limiting ROS production). In turn, iron supplementation appears harmful both in the initial phase4,38 and in further promoting atherosclerosis.23
Lastly, the positive effects of intravenous iron administration cannot be understated, as both the American College of Cardiology and the European Society of Cardiology endorse its use where clinically applicable, such as in absolute or relative iron deficiency in patients with HF.15,99
Conflicts of interestThe authors have no conflicts of interest to declare.
This work was supported by grants from “Iuliu Hațieganu” University of Medicine and Pharmacy, Cluj-Napoca, Romania (Grant No. 1530/35/18.01.2019), and European Social Found, Human Capital Operational Program 2014-2020 (Grant No. POCU/380/6/13/125171).
