






Dilated cardiomyopathy (DCM) is a myocardial disease that can progress to a terminal stage, requiring heart transplantation. In this work we aim to contribute to knowledge of genetic variants in adult patients undergoing heart transplantation due to end-stage DCM, reporting the results obtained in our single-center tertiary hospital series using target next-generation sequencing (NGS).
Methods and ResultsGenetic variants were screened in 15 genes, preselected based on variants previously identified in DCM patients. Thirteen unrelated patients were included, nine (69%) male, mean age at diagnosis 33±13 years, eight (62%) with familial DCM. Nine genetic variants were identified in six (46%) patients: five in LMNA, two in LBD3, one in TNNT2 and one in TCAP. These variants were new in most patients. The majority were classified as of uncertain significance. Two patients were double and triple heterozygotes in the LBD3 and LMNA genes, respectively.
ConclusionOur results highlight the potential of NGS in the genetic characterization of DCM patients. LMNA is one of the most frequently mutated genes and should be included in all target gene assessments of end-stage DCM patients until more data are available.
A miocardiopatia dilatada é uma doença miocárdica que pode evoluir para um estádio terminal, requerendo transplante cardíaco. Neste trabalho, pretendemos contribuir para o conhecimento das variantes genéticas presentes em pacientes adultos submetidos a transplante cardíaco, descrevendo os resultados obtidos utilizando técnicas de sequenciação de ADN de nova geração.
Métodos e resultadosVariantes genéticas foram pesquisadas em 15 genes pré-selecionados com base em variantes previamente identificadas em pacientes com miocardiopatia dilatada. Foram incluídos 13 pacientes não relacionados, nove (69%) do sexo masculino, com idade média na altura do diagnóstico de 33±13 anos, oito (62%) com doença familiar. Foram identificadas nove variantes genéticas em seis (46%) pacientes: LMNA-5, LBD3-2, TNNT2-1 e TCAP- 1. A maioria das variantes genéticas foi classificada como de significado incerto. Dois pacientes eram heterozigotos duplos e triplos nos genes LBD3 e LMNA, respetivamente.
ConclusõesOs nossos resultados reforçam o potencial das novas tecnologias de sequenciação na caracterização genética de doentes com miocardiopatia dilatada. Até que mais dados estejam disponíveis, o gene LMNA é um dos mais frequentemente envolvidos e deverá ser incluído na avaliação de pacientes com miocardiopatia dilatada em fase terminal.
Dilated cardiomyopathy (DCM) is a myocardial disease characterized by left ventricular (LV) dilatation and systolic dysfunction.1 In a subgroup of DCM patients, the disease progresses to a terminal stage, requiring heart transplantation (HT), despite current pharmacological and device therapies. This unfavorable progression may be due to various factors, such as an aggressive initial toxic insult, initiation of therapy late in the course of the disease, or, especially, the presence of an adverse genetic background or of harmful genetic interactions with environmental factors. Based on family screening studies, the percentage of inheritable disease (causality attributed to genetic factors) is estimated to be over 30-50% in so-called idiopathic cases.2 Examples of deleterious variants associated with sudden cardiac death or evolution to terminal DCM have been particularly reported in the LMNA gene.3 Bearing these issues in mind, it is important to explore the molecular mechanisms involved in failure to respond to therapy, knowledge of which is necessary to develop further alternative therapeutic strategies in heart failure syndromes. Until now, assessment of DCM genetics has been hampered by genetic heterogeneity between families, with more than 50 different genes associated with DCM4 and with most of the known genes, except titin, accounting for fewer than 10% of cases.5 The recent development of next-generation sequencing (NGS) techniques has enabled identification of a large and growing number of genetic variants in DCM patients,6 but the major challenge with this technology is the establishment of causal relationships between identified genetic variants and cardiomyopathy phenotypes.7 Recently, the European Society of Cardiology's working group on myocardial and pericardial diseases recommended the use of NGS with very large panels of genes only when the family structure permits segregation analysis.1
Few studies to date have specifically assessed the genetic background of cohorts of HT recipients due to DCM,3,8,9 and even fewer have used NGS approaches.10 Such studies are essential to provide data on the clinical utility of genetic tests in non-responsive DCM patients.
In this work we aim to contribute to knowledge of genetic variants in adult patients undergoing HT due to end-stage DCM, reporting the results obtained in our single-center tertiary hospital series and using target NGS.
MethodsPopulationThe study cohort comprised HT recipients with end-stage DCM (all with left ventricular ejection fraction [LVEF] <30%) from a single tertiary referral hospital.
The study is part of a larger Portuguese project which aims to collect clinical and molecular data on adult patients with DCM.11 DCM was classified as familial when idiopathic disease was present in more than one family member or when unexplained sudden cardiac death occurred under the age of 35 in at least one first-degree.
General clinical assessment methods, such as electrocardiogram, echocardiogram and 24-hour Holter monitoring, have been previously reported.11 In this patient subgroup, cardiac catheterization was performed in all cases.
Next-generation sequencingAll patients provided a peripheral blood sample for molecular analysis. The samples were sent to a central laboratory, where DNA was extracted for molecular analysis and stored at -70°C.
Genetic variants in 15 genes – LMNA/C, MYH7, MYBPC3, TNNT2, ACTA1, TPM1, CSRP3, TCAP, SGCD, PLN, MYL2, MYL3, TNNI3, TAZ and LDB3 – were screened in all samples, using polymerase chain reaction (PCR) with direct sequencing (NGS with at least 30-fold coverage combined with Sanger sequencing). Genes were preselected based on genetic variants previously identified in DCM patients.
Primers were designed for all coding exons, covering exon-intron boundaries using the open-source software Primer3. No known variants were present in the primer sequences (dbSNP build 130). A multiplex PCR-based method was used to reduce the number of amplification reactions (primer sequences can be provided on request). Multiplex PCR reactions were performed following the QIAGEN Multiplex PCR Kit protocol (QIAGEN, Hilden, Germany).
Library and template preparationThe quality of patient genomic DNA samples was evaluated by gel electrophoresis and quantification was performed using the Qubit dsDNA HS Assay Kit (Life Technologies). A total of 50 ng of genomic DNA was used in each multiplex PCR reaction. Libraries were prepared using the Ion Xpress™ Plus gDNA protocol and Amplicon Library Preparation (part number 4471989 Rev. C) (Life Technologies). Libraries were quantified using the Qubit dsDNA HS Assay. All libraries were diluted to the same concentration and pooled to ensure equal representation of the different samples. The diluted and combined libraries underwent amplification by emulsion PCR using the Ion PI™ Template OT2 200 Kit (Life Technologies) on an Ion OneTouch 2 Instrument (Life Technologies) according to the manufacturer's instructions. Ion Sphere particles were enriched using the Ion OneTouch 2 enrichment system (Life Technologies).
Semiconductor sequencing and data analysisSequencing was carried out on an Ion PGM system using semiconductor technology. The Ion Sequencing Kit, version 2.0 (Life Technologies) was used to perform sequencing runs, following the manufacturer's recommendations. Data from the PGM runs were processed using the platform-specific Ion Torrent Suite pipeline software, version 4.2 (Life Technologies) to generate sequence reads, trim adapter sequences, filter and remove poor signal reads, and split the reads according to the barcode. Reads were assembled with SeqMan NGen, version 4.1 (DNASTAR, Madison, WI) using the FASTQ files containing sequence reads, and the template references were adjusted for the covered amplicons. SeqMan Pro version 10 (DNASTAR) was used for post-assembly analysis of the overall amplicon coverage, individual base depth of coverage, and variant identification.
Pathogenicity was assessed by comparing the results with databases in which genetic variants had been reported, as well as using predictive models and segregation analysis, in accordance with the guidelines of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.12
Subsequently, the patients’ relatives were screened for the identified genetic variants. Clinical assessment, electrocardiogram and echocardiogram were performed in all available family members.
EthicsInformed consent was obtained from all patients and relatives. The study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki as confirmed by prior approval by the institution's human research committee.
ResultsClinical characteristicsThirteen unrelated patients were included, nine (69%) male, with a mean age at DCM diagnosis of 33±13 years, eight (62%) with familial DCM.
Native heart LVEF was 18.2±5.8% and LV end-diastolic diameter was 67.5±10.2 mm. Five patients (38%) presented with left bundle branch block and three were in non-sinus rhythm, two with atrial standstill. Cardiac magnetic resonance imaging was performed in four patients (31%) and endomyocardial biopsy in two, which revealed non-specific alterations. Elevation of serum creatine kinase in at least one laboratory assessment was detected in three (23%) cases, in two of which neuromuscular assessment confirmed the presence of skeletal myopathy (Table 1). Seven patients (54%) had received an implantable cardioverter-defibrillator (ICD) and three (23%) an LV resynchronization device before HT.
Clinical characteristics of the study population.
| Patient | Gender/age, years | ECG before HT | Native heart LVEF, %/LVED, mm | Comments | Genetic variant identified |
|---|---|---|---|---|---|
| 1 | F/47 | SR/- | 15/65 | Postpartum DCM diagnosis | No |
| 2 | M/55 | SR/LBBB | 8/87 | Yes | |
| 3 | M/44 | SR/LVH | 29/55 | CK elevation, no evidence of skeletal myopathy; transmural apical and lateral LE on CMRI; no evidence of MI on explanted heart tissue | Yes |
| 4 | M/60 | SR/LBBB | 15/78 | Intramyocardial mid-septal LE in on CMRI | Yes |
| 5 | M/56 | Atrial flutter/LBBB | 16/63 | No | |
| 6 | M/53 | Atrial standstill/RBBB+LAH | 10/59 | CK elevation with statins, limb-girdle myopathy, skeletal biopsy with lobulated fibers | No |
| 7 | M/36 | SR/LVH | 23/70 | Yes | |
| 8 | F/32 | SR/- | 16/59 | No LE on CMRI | No |
| 9 | M/61 | SR/RBBB+LAH | 23/67 | No | |
| 10 | M/60 | Atrial standstill/ventricular pacing | 24/58 | CK elevation, rigid spine syndrome | Yes |
| 11 | F/26 | SR/LVH | 17/83 | No LE on CMRI | No |
| 12 | M/47 | SR/LBBB | 21/60 | Yes | |
| 13 | F/53 | SR/LBBB | 20/74 | No |
CK: creatine kinase; CMRI: cardiac magnetic resonance imaging; DCM: dilated cardiomyopathy; ECG: electrocardiogram; HT: heart transplantation; LAH: left anterior hemiblock; LBBB: left bundle branch block; LE: late enhancement; LVED: left ventricular end-diastolic diameter; LVEF: left ventricular ejection fraction; LVH: left ventricular hypertrophy; MI: myocardial infarction; RBBB: right bundle branch block; SR: sinus rhythm.
We were able to assess 35 relatives of eight of the probands, of whom six (17%) presented DCM and two (6%) previously documented LV systolic dysfunction, from which they recovered (reverse remodeling).
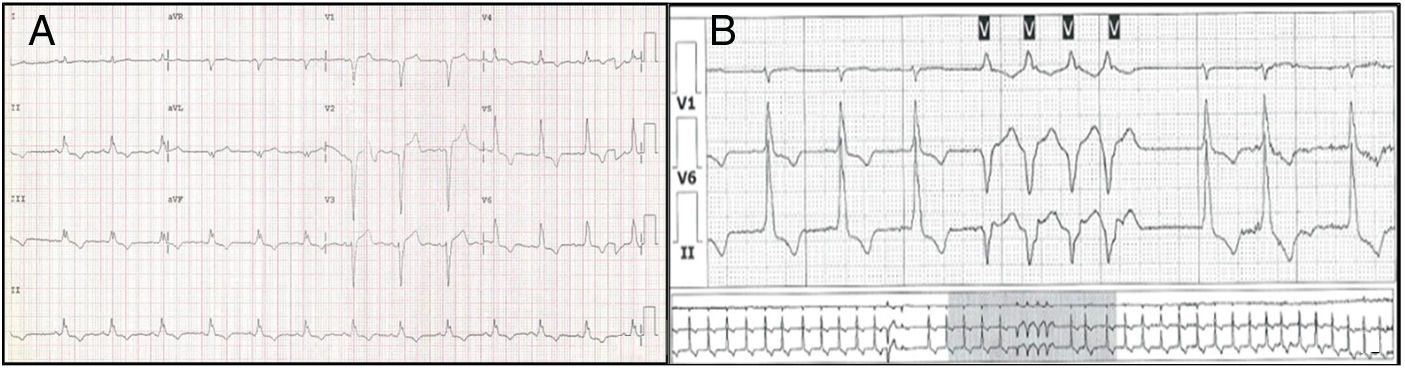
Genetic analysisNine genetic variants were identified in six (46%) patients: five in LMNA, two in LBD3, one in TNNT2 and one in TCAP. These variants were new in most patients. Two patients (15%) were double and triple heterozygotes in the LBD3 and LMNA genes, respectively. The majority were classified as of uncertain significance. Only one variant, in LMNA, was classified as likely pathogenic. This variant, in exon 6 of LMNA (c.1003C>T), leads to an amino acid substitution at position 335 of the protein (p.Arg335Trp) and has been reported in other DCM patients.13Figures 1-3, respectively, show the baseline ECG, pathologic features of the explanted heart, and the genogram of patient 3, in whom this variant was identified. Genetic family screening enabled familial non-segregation of the TCAP variant to be established. Table 2 depicts the final classification of the genetic variants detected in the present study.
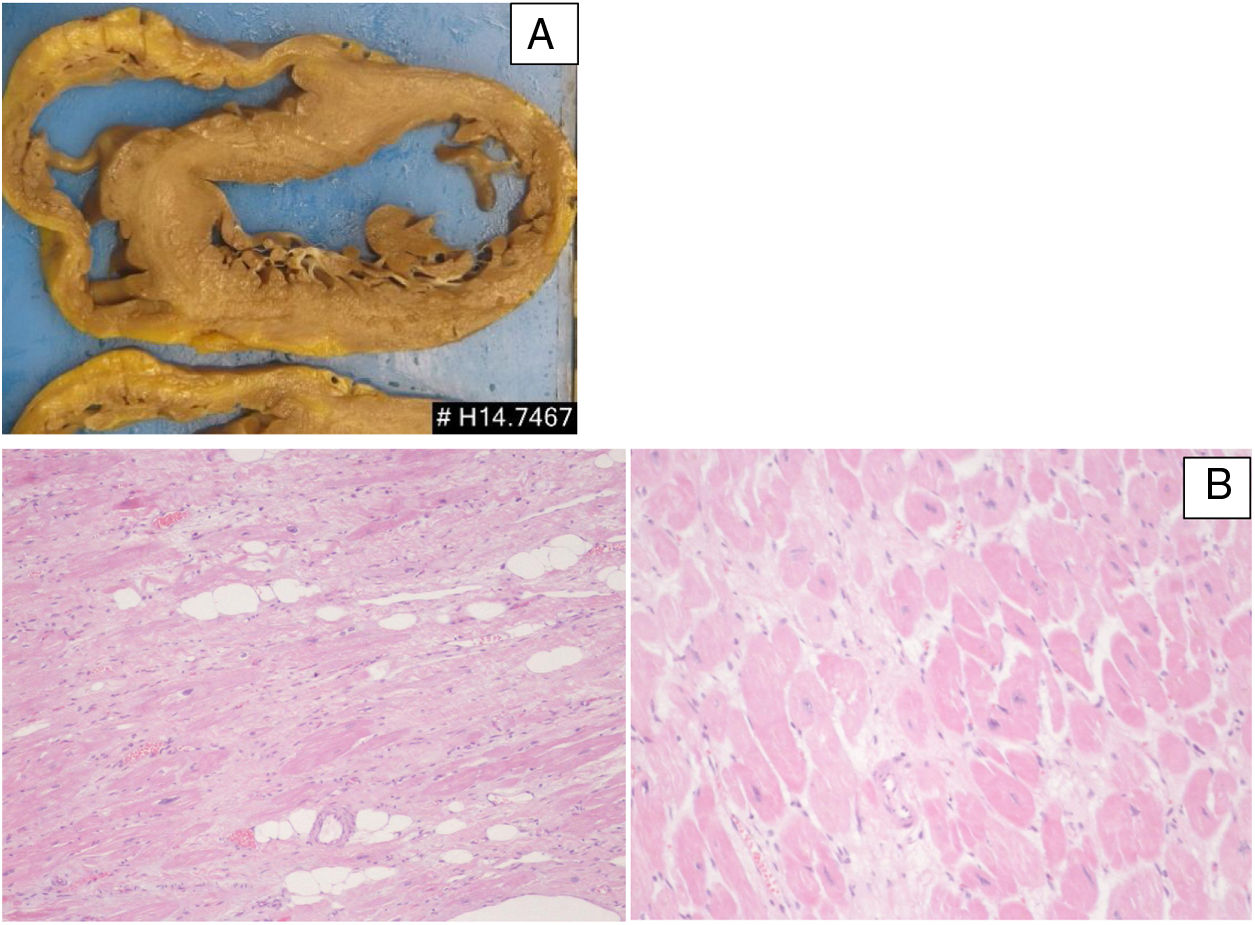
Patient 3: pathologic features of the explanted heart. (A) Macroscopy of heart section revealing features of dilated cardiomyopathy; (B) microscopic features (H&E stain) revealing interstitial fibrosis, hypertrophic cardiomyocytes and scattered adipocytes (left: low power; right: high power).
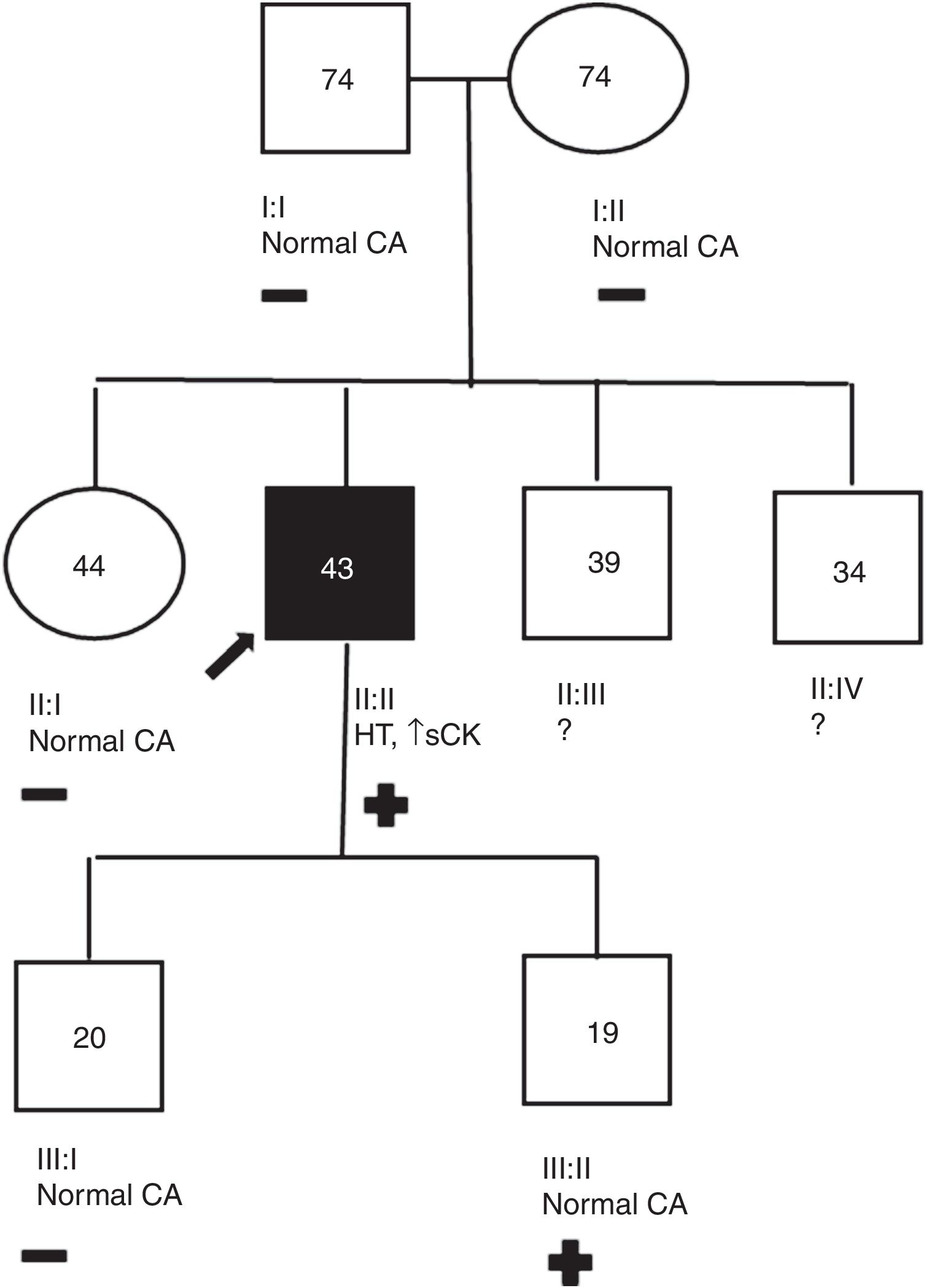
Patient 3 genogram. Square: male; circle: female; dark symbol: dilated cardiomyopathy; +/-: presence/absence of LMNA c.1003C>T variant; numbers inside symbols: current age (years); ↑ sCK: elevated serum creatine kinase; CA: cardiac assessment; HT: heart transplantation; ?: unavailable for clinical/genetic assessment.
Classification of genetic variants detected (in accordance with the guidelines of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology12).
| Patient | Gene | NM (NCBI) | Variant | Final classification (ACMG) |
|---|---|---|---|---|
| 310101012 | LMNA | NM_170707.3NM_170707.3NM_170707.3NM_170707.3NM_170707.3 | c.1003C>T; p.Arg335Trpc.1318G>A; p.Val440Metc.1604G>C; p.Gly535Alac.1982G>C; p.Cys661Serc.460G>A; p.Glu154Lys | Likely pathogenicVUSVUSVUSVUS |
| 44 | LBD3 | NM_007078.2NM_007078.2 | c.466G>A; p.Ala156Thrc.1189G>A; p.Val397Ile | VUSVUS |
| 7 | TNNT2 | NM_001001430.2 | c.325C>T; p.His109Tyr | VUS |
| 2 | TCAP | NM_003673.3 | c.313G>C; p.Glu105Gln | VUS |
ACMG: American College of Medical Genetics and Genomics; NCBI: National Center for Biotechnology Information; VUS: variant of uncertain significance.
DCM is a major indication for HT, and a lower LVEF indicates a higher risk for this therapeutic option.14 However, HT is not necessarily a definitive treatment, due to complications arising from graft failure and chronic immunosuppression. The median life expectancy of transplant patients is still around 15 years.15 At the same time, insufficient donor hearts are available for all candidates and many patients die while on the waiting list. Given this situation, it is essential to further explore the mechanisms behind failure to respond to heart failure therapies in patients who progress to end-stage disease.
Genetics appears to explain a significant proportion of DCM cases. However, in most HT recipients, due to the scarcity of specific etiologic markers and the fact that genetic alterations are not systematically analyzed in these patients, DCM is frequently categorized as idiopathic. Systemic manifestations can occasionally alert the clinician to the possibility of a genetic disease, such as the presence of skeletal myopathy or atrioventricular block in carriers of LMNA variants.
The recently developed NGS technologies are more effective in detecting genetic variants in HT candidates, but the most efficient way to use NGS in clinical practice remains to be established, in particular how to select the panel of genes to be included in a target gene approach.
The few reports of genetic yields obtained specifically in HT cohorts of DCM patients use different approaches. Initial studies with Sanger DNA sequencing revealed a relatively high frequency of LMNA variants,3 while more recently, Cuenca et al., after screening a significant number of genes by NGS, reported pathogenic variants in 40% of patients, the most frequent in emerin and titin genes.10
In our study, genetic variants were a common finding (46%), but only one variant was categorized as likely pathogenic, due to the small and non-informative families. We also found a high percentage of LMNA variants (23%), followed by variants in LBD3 and TNNT2. The patient with the pathogenic LMNA variant presented elevated serum creatine kinase, but this observation was only detected once, and has not been associated with overt clinical myopathy. This missense variant, located in the coil 2 domain of the lamin A protein, has also been reported in another DCM patient with no evidence of muscular dystrophy, who required an ICD due to ventricular tachycardia and underwent HT at the age of 39.13
Two other patients presented stable rigid spine syndrome and limb-girdle myopathy, respectively, but in neither case did skeletal involvement limit HT. These cases highlight the well-known importance of neuromuscular assessment in DCM patients.16
Concerning the presence of two or more genetic variants in the same individual, it is not known if or how this may modify the phenotype. Double heterozygosity for LMNA and titin variants has been shown to be associated with a more severe clinical course, in terms of age of terminal heart failure and HT in a DCM family.17 Compound and digenic heterozygosity also appear to be common among hypertrophic cardiomyopathy (HCM) patients who develop end-stage heart failure.18 Of note, it is possible that end-stage DCM may represent end-stage HCM, but this is less likely in our patients, given the follow-up before HT, and since histopathology of the explanted hearts did not reveal myofibril disarray in any of the cases. On the other hand, the lack of segregation of genetic variants does not mean they have no biological effects, particularly when detected in family members with more severe phenotypes.
Major limitations of our study are the small number of patients and the limited number of studied genes, especially the fact that we did not include titin, in which genetic variants appear to have an important role in the pathogenesis of DCM.19
ConclusionsThe application of NGS may help to expand our knowledge of the genetic mechanisms of cardiomyopathies that do not respond to medical therapy, including in DCM patients undergoing HT. It is important to clarify the pathogenicity of identified variants, and until more data are available, most variants will be classified as of uncertain significance, as in our patients. The impact of compound or digenic heterozygosity should also be investigated, in view of the growing number of clinical and genetic reports of patients with such mutations.
LMNA is one of the most frequently mutated genes in HT patients and should be included in all target gene assessments of end-stage DCM patients.
Conflicts of interestThe authors have no conflicts of interest to declare.
This study received a research grant from Fundação para a Ciência e Tecnologia (FCT-PTDC/BIM-MEC/0650/2012).
