








Atualmente, o diagnóstico da Síndrome de Brugada (SB) obriga à documentação do padrão eletrocardiográfico de repolarização característico denominado tipo 1. Em 38% dos casos familiares desta entidade encontra-se uma mutação do gene SCN5A responsável pela síntese do canal de sódio NaV1.5. A disfunção desta corrente de sódio repercute-se no potencial de ação cardíaco de resposta rápida, fundamentalmente nos miócitos auriculares, nas fibras de Purkinje e nos miócitos ventriculares.
ObjetivosDetetar portadores de mutação do SCN5A sem recorrer ao padrão de repolarização no ECG.
MétodosA partir de 3 famílias e de um total de 141 elementos, dos quais 55 portadores (P+) de duas mutações non-sense específicas do SCN5A causadoras de SB, foram estudados os maiores de 16 anos (113 elementos/42 P+). Foi medido no ECG o intervalo PR (PR) em DII e dispersão QT (dQT) entre V1 e V3. No ECG de alta resolução (ECGAR) determinado o QRS filtrado (QRSf), root-mean square (RMS40) e low-averaged signal (LAS). Para detetar P+ foi criado: 1) um teste de rastreio (TesteRas) constituído pelo intervalo PS (PR+QRSf) ≥ 250 (250ms corresponde a 80% do valor teórico máximo deste intervalo em saudáveis) e 2) um teste diagnóstico (TesteDx) que resulta do cumprimento simultâneo de 4 condições: PS ≥ 250 e dQT > 10 e LAS ≥ 26 e RMS40 ≤ 29 (estes 2 últimos valores correspondem aproximadamente a 70% do valor teórico máximo em saudáveis).
ResultadosEncontradas diferenças significativas entre os P+ versus não portadores (P-) do PR, da dQT, do QRSf, do LAS e da RMS40. O gene está associado a todas estas variáveis, sendo a associação mais forte com o PR. A 63 elementos (38 P+) foi aplicado o TesteRas e o TesteDx. O TesteRas foi positivo em 38 de 38 P+ com 8 falsos positivos em 27 P- (sensibilidade [S] =100% e especificidade [E] =66,67%). Na curva Roc para este teste o cut-off PS=252,5 tem S=100% e E=76% e o cut-off PS=260 tem S=94,7% e E=84%. O TesteDx foi positivo em 36 de 38 P+ com 3 falsos positivos: S=94,74% e E=88,89%. Por modelo logístico multivariado identifica-se um cut-off através da curva Roc com S=92% e E=92%. A S e E de diagnóstico do ECG espontâneo usando o padrão de repolarização tipo 1 no mesmo grupo de doentes foram de 52,4% e 97,2%, respetivamente (a diferença de S entre o TesteDx e o padrão de repolarização no ECG é estatisticamente significativa).
ConclusõesO TesteRas e o TesteDx são uma ferramenta mais eficaz do que o padrão de repolarização típico para distinguir entre P+ e P- destas duas mutações non-sense do SCN5A. Sugerimos a sua utilização em familiares diretos de casos índice com SB, quando o resultado genético (ainda) não está disponível e num score de probabilidade de doença em indivíduos com ECG de Brugada idiopático ou em indivíduos com sintomas relacionados com arritmias e padrão de repolarização tipo 2 ou 3 de Brugada.
Diagnosis of Brugada syndrome (BS) currently requires documentation of a characteristic repolarization pattern (type 1 Brugada ECG). Mutations in the SCN5A gene, which codes for sodium channel NaV 1.5, are found in 38% of familial cases of BS. Sodium current dysfunction negatively affects the cardiac fast response action potential, particularly in atrial and ventricular myocytes and in the fast-conducting Purkinje system.
ObjectivesTo detect carriers of SCN5A mutations without using the characteristic repolarization pattern (type 1 Brugada ECG).
MethodsOf a total of 141 members of three different families including 55 carriers of two nonsense SCN5A mutations causing BS, all those aged over 16 (113 individuals, 42 carriers) were studied. The PR interval (PR) and QT dispersion (QTd) between leads V1 and V3 were measured on conventional ECG. Using signal-averaged ECG the total duration of the filtered QRS complex (fQRS), the root-mean-square (RMS40) and the low-amplitude signal (LAS) were measured. The following procedures were developed to detect carriers/To detect carriers the following procedures were developed: (1) a screening test (ScreenTest) with PS (PR+fQRS) ≥250 (250ms is 80% of the theoretical maximum in healthy individuals); and (2) a diagnostic test (DiagTest) for the simultaneous fulfillment of four conditions: PS ≥250 and QTd ≥10 and LAS >26 and RMS40 ≤29 (the latter two cut-offs are approximately 70% of the theoretical maximum in healthy carriers).
ResultsSignificant differences in PR, QTd, QRSf, RMS40 and LAS were found between carriers and non-carriers. The SCN5A gene was associated with all these variables, the strongest association being with PR. Both tests were applied to 63 family members (38 carriers). The ScreenTest was positive in 38 of 38 carriers, with eight false positives in 27 non-carriers (sensitivity [SE]=100% and specificity [SP]=66.67%). From ROC curve analysis a cut-off of PS=252.5 shows SE=100% and SP=76% and a cut-off of PS=260 shows SE=94.7% and SP=84%. The DiagTest was positive in 36 of 38 carriers, with three false positives: SE=94.74% and SP=88.89%. From ROC curve analysis a multivariate logistic model identifies a cut-off with SE=92% and SP=92%. In the same group the SE and SP of the characteristic spontaneous repolarization pattern (type 1 Brugada ECG) to detect carriers were 52.4% and 97.2%, respectively, and the difference between the SE of the DiagTest and of the typical repolarization pattern is statistically significant.
ConclusionsThe ScreenTest and DiagTest are more effective tools than the characteristic repolarization pattern to discriminate between carriers and non-carriers of these two nonsense SCN5A mutations. We suggest their use in first-degree relatives of Brugada patients when the results of genetic testing are not available, in a score of disease probability in individuals with idiopathic Brugada ECG, and in patients with arrhythmias or other Brugada-related symptoms presenting type 2 or type 3 Brugada ECG.
A Síndrome de Brugada (SB) foi descrita como uma nova entidade pela primeira vez em 1992, caracterizando-se por um padrão eletrocardiográfico típico (bloqueio do ramo direito do feixe de His e supradesnivelamento do segmento ST nas derivações precordiais direitas) ao qual se associa uma predisposição aumentada para arritmias ventriculares malignas e morte súbita em indivíduos sem cardiopatia estrutural1. Em menos de duas décadas passou de uma mera curiosidade médica a uma entidade que é obrigatório diagnosticar, estratificar e tratar, pois estima-se que seja responsável por 20 a 50% dos casos de morte súbita nos indivíduos com coração estruturalmente normal2, acrescido o facto de a idade média do diagnóstico e/ou de o evento que o promove (que é muitas vezes a morte súbita) ser aos 40±22 anos. Pertence ao grupo das canalopatias, doenças que resultam de disfunção primária dos canais responsáveis pelo potencial de ação cardíaco, predispondo e/ou perpetuando arritmguraias sem doença estrutural concomitante3.
Perspetiva históricaÉ uma doença congénita, com transmissão familiar frequente. A primeira mutação associada foi descrita em 1998 no gene SCN5A que codifica a subunidade α do canal de sódio cardíaco, o NaV1.54 e, a partir daí, mais de 100 mutações diferentes neste gene foram publicadas5. De salientar que estas diferentes mutações podem ser classificadas como non-sense ou missense e não têm todas o mesmo impacto fisiopatológico. As primeiras são habitualmente mais graves, pois originam uma proteína truncada e disfuncional, porque mais curta ou mais longa que a proteína normal. As mutações missense levam à substituição de um aminoácido, mas a proteína permanece completa. Nestes casos, a consequência biológica sobre a função da proteína é incerta e são necessários estudos funcionais in vitro para apurar a sua relevância.
O SCN5A mutado pode acarretar perda de função da corrente de sódio (INa) no potencial de ação cardíaco, seja por défice de expressão quantitativa ou por disfunção qualitativa do canal; porém, as mutações são encontradas em apenas 18 a 30% dos doentes2 e em 38% dos casos familiares6. Esta baixa incidência pode ser justificada por heterogeneidade genética1 com outros genes já descritos e associados à SB (presumivelmente haverá muitos outros ainda por descrever): (1) o glicerol-3-fosfato desidrogenase 1-like (GPD-1L) é responsável pelo trânsito do NaV1.5 para a superfície celular e, quando mutado, causa SB por redução em 31% da quantidade de canais disponíveis na membrana e por 50% de redução da corrente de sódio7, (2) mutações do canal de cálcio cardíaco (CACNA1c e CACNB2b) associadas a uma entidade de sobreposição: síndrome de QT curto com padrão de Brugada no ECG8 e (3) mutações do gene KCNE3, que é um modelador da função do canal KV4.3 que é responsável pela corrente transitória de saída de potássio (ITO)9. Todas estas mutações independentes do gene SCN5A mais recentemente descritas obrigaram ao abandono do paradigma inicial da SB como uma canalopatia do sódio «pura» para o conceito atual mais abrangente de canalopatia da fase 1 do potencial de ação cardíaco, por desequilíbrio de uma das várias correntes iónicas (sódio, potássio e cálcio) que nele participam, seja por mutação de genes de canais iónicos ou por mutação de genes de proteínas associadas à atividade ou ao transporte dos mesmos10.
Diagnóstico da SBAtualmente, o diagnóstico da SB é definitivo apenas quando se observa o ECG de tipo 1 em pelo menos 2 derivações precordiais direitas, na presença ou ausência de um bloqueador dos canais de sódio, em associação com um ou mais dos seguintes: fibrilhação ventricular (FV) documentada, taquicardia ventricular (TV) documentada, história familiar de morte súbita cardíaca (< 45 anos), ECG de tipo 1 em elementos da família, TV/FV por estimulação elétrica programada em estudo eletrofisiológico (EEF), síncope ou respiração noturna agónica2.
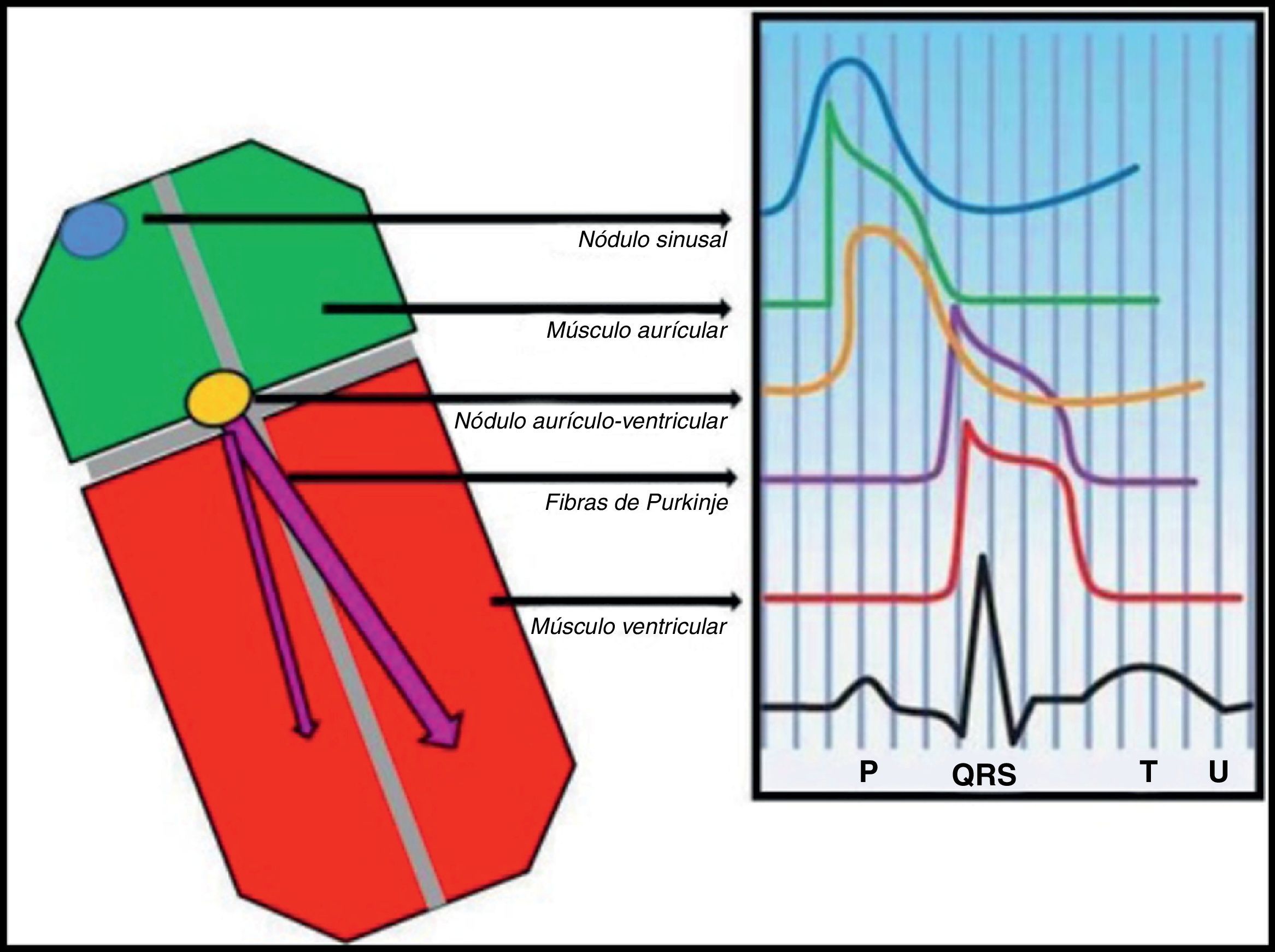
Do gene ao eletrocardiogramaExistem 2 teorias para explicar a manifestação eletrocardiográfica da SB: (1) atraso da condução no epicárdio da parede livre do trato de saída do ventrículo direito ou (2) repolarização prematura do potencial de ação do epicárdio do ventrículo direito ou uma combinação de ambas. Na primeira, a ativação assíncrona do epicárdio em relação ao endocárdio por disfunção de um canal (NaV1.5 ou outro) cria um gradiente de voltagem entre estas camadas de células. Na segunda (Figura 1), a disfunção de um canal (NaV1.5 ou outro) com desequilíbrio na fase 1 a favorecer a repolarização por desproporção iónica a favor da corrente ITO (saída de cargas positivas) leva a que a membrana repolarize para além da voltagem a que os canais de cálcio tipo-L são ativados, falhando a fase 2 do potencial de ação. Em ambos os modelos é gerado um gradiente de voltagem transmural entre o epicárdio e o endocárdio que se manifesta no ECG como uma elevação em cúpula do segmento ST ≥ 2mm (0,2mV) seguido por onda T negativa (ECG tipo 1 de Brugada) e que é um substrato arrítmico para reentradas de fase 2. Em ambas as hipóteses o atingimento preferencial do trato de saída do ventrículo direito é explicado pelo facto de a corrente ITO ter maior expressão funcional no epicárdio desta região. Estas alterações da repolarização no ECG que levam ao diagnóstico da SB traduzem disfunção do potencial de ação cardíaco de resposta rápida ventricular. O mesmo potencial de ação é também responsável pela despolarização dos miócitos auriculares e das fibras de Purkinje além do ventrículo (Figura 2)11,12. Assim, podemos teorizar que a disfunção do potencial de ação responsável pelas perturbações da repolarização descritas pode também resultar em aumento ou até perturbação (sub-) clínica da condução intra-auricular, aurículoventricular e intraventricular.
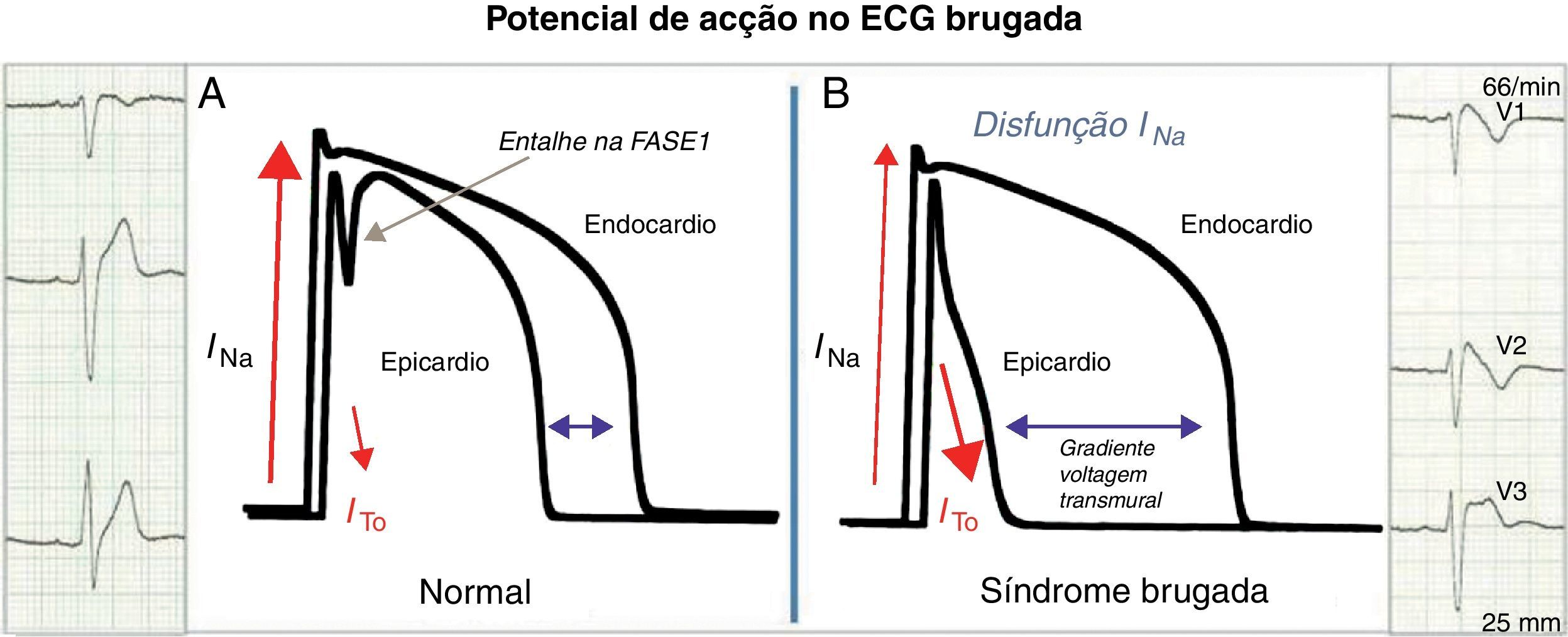
A Nos saudáveis, o potencial de ação dos miócitos ventriculares apresenta um pequeno entalhe na fase 1 mediado pela corrente ITO (mais evidente no epicárdio) sem tradução no ECG. B Na SB, o desequilíbrio iónico na fase 1 a favorecer a repolarização no epicárdio forma um gradiente de voltagem transmural que se traduz no ECG como o padrão de repolarização tipo 1.
O diagnóstico da SB é evidente e elementar na sua forma de apresentação mais característica, como um doente com o padrão de repolarização do tipo 1 espontâneo nas derivações precordiais direitas e com episódio de paragem cardíaca abortada. O padrão de repolarização do tipo 1 sem qualquer outro critério deve ser descrito como ECG de Brugada idiopático, não como SB, e a sua abordagem clínica é alvo de grande controvérsia13. Neste caso, deve ser sempre considerada uma extensa lista de diagnósticos diferenciais que podem mimetizar as alterações do ECG (Tabela 1)5; porém, é necessário ter em conta que aguardar pelo primeiro sintoma para confirmar um diagnóstico na SB pode ser catastrófico: em 77% dos doentes a paragem cardíaca é a primeira manifestação da doença.
Situações clínicas que podem mimetizar o padrão de repolarização de Brugada
| Bloqueio do ramo direito do feixe de His atípico |
| Enfarte agudo do miocárdio do ventrículo direito |
| Pericardite aguda |
| Hemopericárdio |
| Tromboembolia pulmonar |
| Dissecção da aorta |
| Desordens do sistema nervoso central |
| Distrofia muscular de Duchenne |
| Ataxia de Friedreich |
| Hipertrofia do ventrículo esquerdo |
| Cardiomiopatia arritmogénica do ventrículo direito |
| Compressão mecânica do trato de saída do ventrículo direito (tumor / pectus excavatum) |
| Após cardioversão elétrica |
| Repolarização precoce |
| Hipotermia |
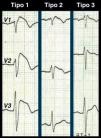
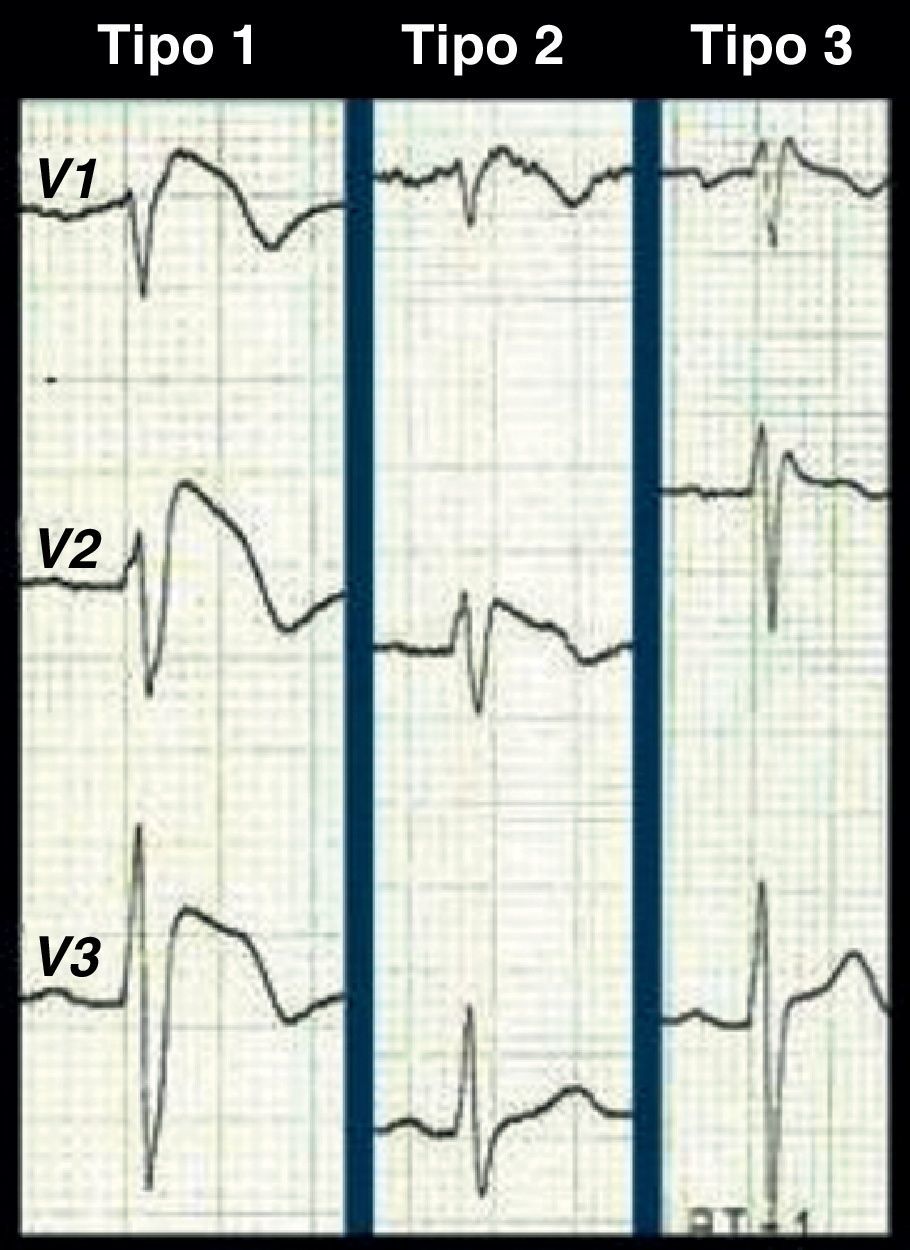
Os problemas de diagnóstico continuam nos casos com padrões de repolarização borderline ou Brugada-like com ou sem sintomas. Além do ECG tipo 1, existem mais 2 padrões de repolarização associados à SB (Figura 3), o tipo 2 (elevação do ponto J≥2mm, segmento ST descendente mas sempre 1mm acima da linha isoelétrica e onda T positiva ou bifásica, em sela de cavalo) e o tipo 3 (elevação do segmento ST<1mm a partir do ponto J≥2mm, em arco, em sela de cavalo ou ambas). Segundo a reunião de consenso para o diagnóstico da SB, os padrões tipo 2 e 3 são apenas sugestivos e não diagnósticos, recomendação que não é seguida por todos os autores13,14. A utilização estrita do padrão tipo 1 ou a utilização dos 3 padrões como diagnóstico tem obviamente repercussão na sensibilidade e na especificidade do ECG em detetar Brugada (maior sensibilidade ao utilizar os 3 padrões, maior especificidade usando apenas um). O padrão de repolarização do tipo 3 é quase indistinguível do padrão de bloqueio incompleto do ramo direito do feixe His usual no jovem: negligenciá-lo pode ter resultados catastróficos mas sobrevalorizá-lo pode servir apenas para estigmatizar um indivíduo saudável a um diagnóstico com um peso pessoal, familiar, social e profissional avassalador. Esta discordância tem alimentado polémicas na literatura médica e presume-se ter «contaminado» algumas bases de dados com indivíduos saudáveis, justificando as grandes assimetrias publicadas relativamente ao prognóstico da entidade15–17.
Após confirmação do diagnóstico em qualquer caso índice, é necessário o estudo de todos os familiares possivelmente implicados com ECG basal e/ou teste de provocação. Para o estudo de familiares potencialmente afetados, o ECG basal é insuficiente, pois 51% dos indivíduos afetados apresenta flutuações entre ECG diagnóstico e não diagnóstico18. O teste de provocação com bloqueadores dos canais de sódio (flecainida, ajmalina, procainamida) é, pois, fundamental para as formas intermitentes e/ou ocultas; porém, tem uma sensibilidade, especificidade e reprodutibilidade que são mal conhecidas19.
ECG de alta resoluçãoO ECG de alta resolução (ECGAR) é uma técnica computorizada de aquisição do ECG de forma a detetar anomalias no QRS subtis e demasiado finas para o ECG convencional. O QRS obtido por este exame (figura 4) é a média aritmética do somatório de múltiplos complexos (habitualmente 200), colhidos em cerca de 5 minutos num ambiente com o mínimo de ruído sonoro possível. O processo aumenta a razão sinal-ruído dos complexos cardíacos colhidos, que permite no QRS final detetar sinais da ordem de grandeza do microvolt (μV) e fazer medições com a precisão de milissegundos (ms), que vulgarmente se designam por pós-potenciais ventriculares20.
Objetivos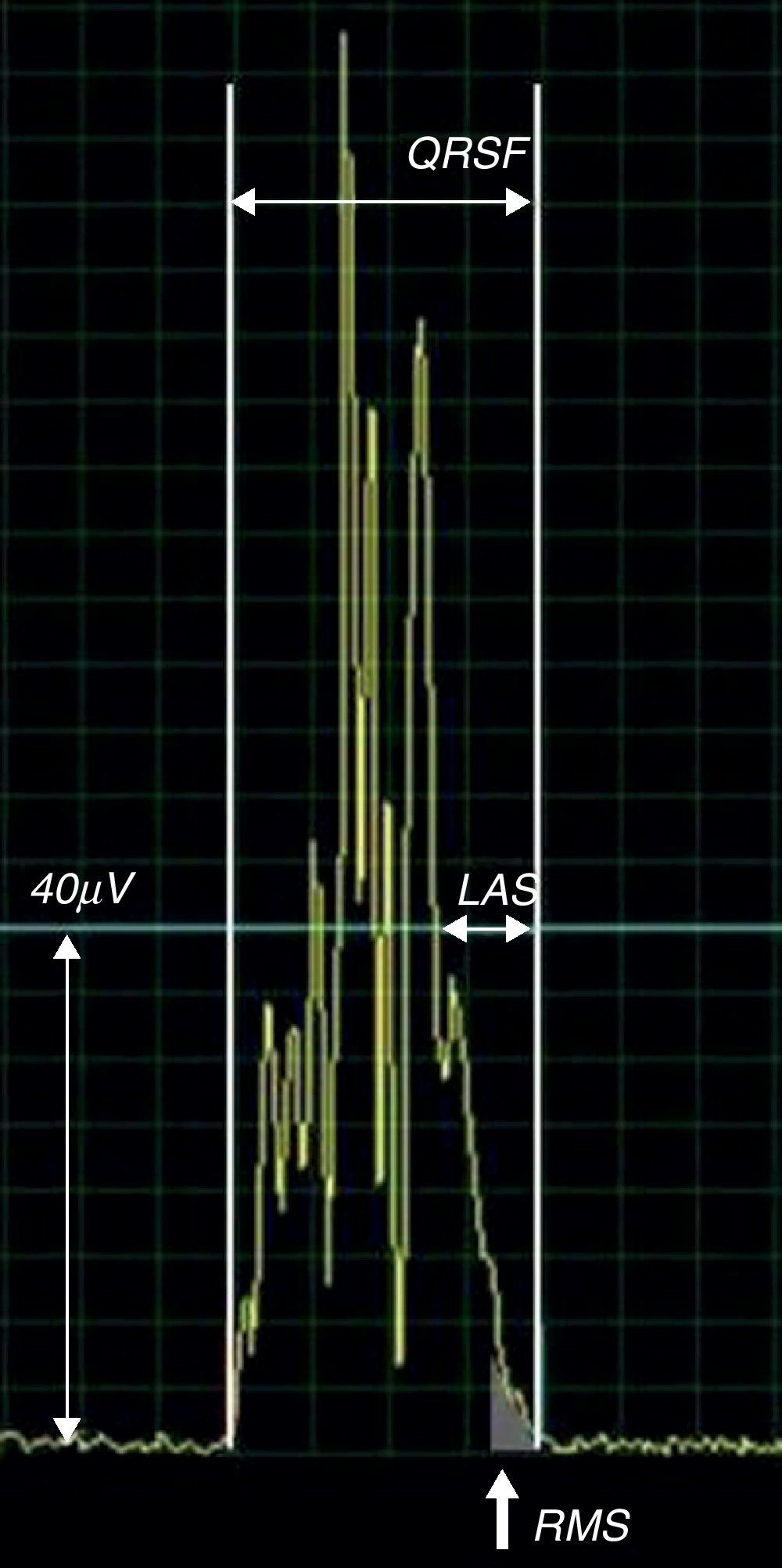
Construir um modelo para detetar portadores de mutações do SCN5A non-sense ou missense com rebate funcional importante baseado também em perturbações subclínicas da condução intra-auricular, aurículoventricular e intraventricular. A partir dele, é construído um score de probabilidade de doença que sugerimos usar em familiares diretos de doentes com SB e em indivíduos com ECG de Brugada idiopático.
MétodosA partir de 3 famílias e de um total de 141 elementos, dos quais 55 portadores de duas mutações non-sense do SCN5A causadoras de SB (43 com Arg222STOP e 12 com Thr1754ProfsX32), foram estudados os maiores de 16 anos.
Foi medido no ECG convencional utilizando calibração 20 mm=1mV e velocidade 50mm/s o intervalo PR (PR) na derivação DII e a dispersão do QT (dQT) entre V1 e V3. Foi realizado o ECGAR com análise no domínio do tempo. Para detetar portadores foram criados 2 testes:
- 1)
Teste de Rastreio (TesteRas) constituído pelo intervalo PS ≥ 250 (em que PS corresponde à soma de PR e QRSf (o valor teórico máximo deste intervalo em saudáveis é 314ms (200+114) e 250ms corresponde a 80% desse valor extremo);
- 2)
Teste Diagnóstico (TesteDx) que resulta do cumprimento simultâneo de 4 condições: PS ≥ 250 e dQT > 10 e LAS ≥ 26 e RMS40 ≤ 29 (estes 2 últimos valores correspondem aproximadamente a 70% do valor teórico máximo em saudáveis).
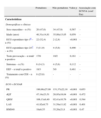
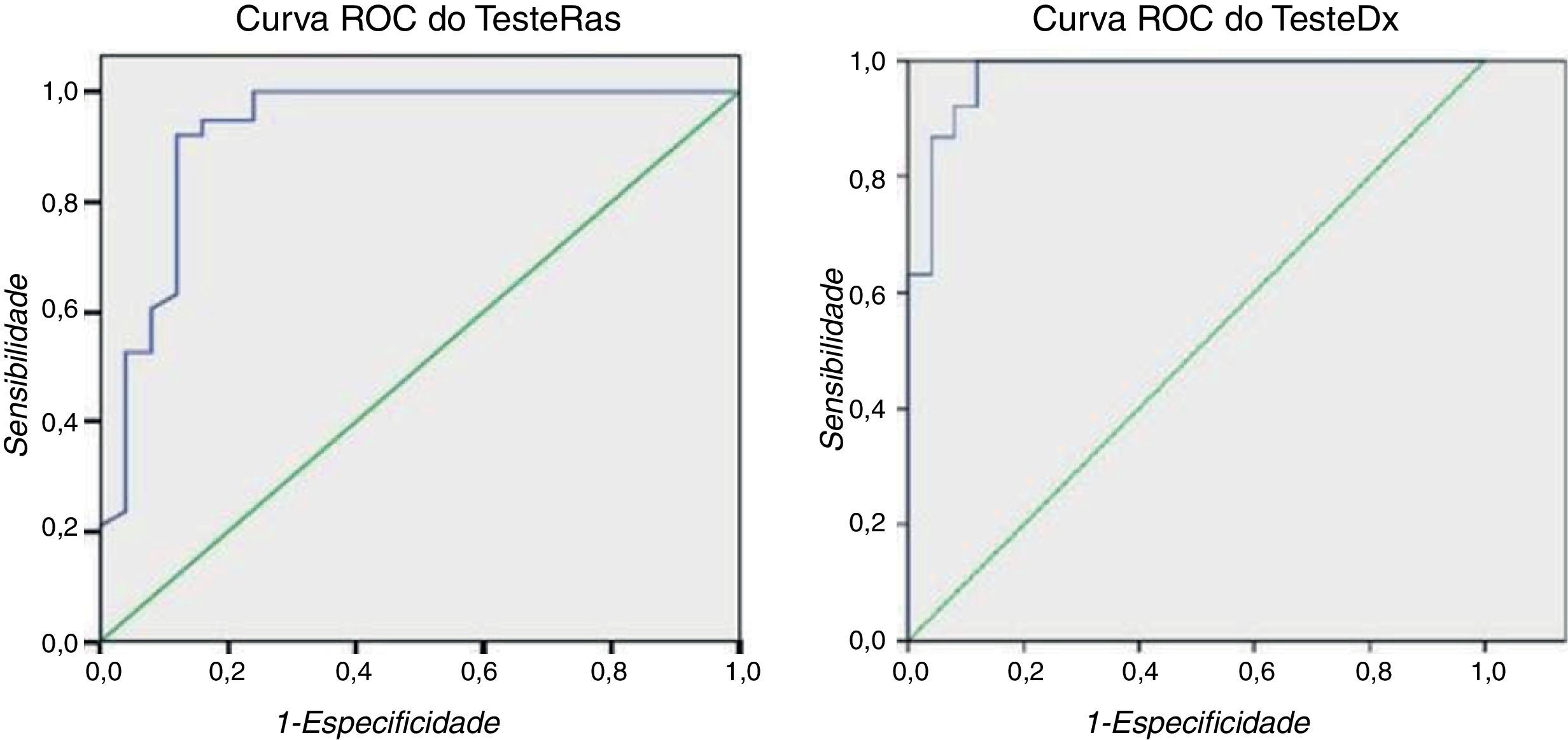
Foram estudados 113 elementos/42 portadores e encontradas diferenças significativas entre os portadores versus não portadores do PR, da dQT, do QRSf, do LAS e da RMS40. O gene está associado a todas estas variáveis, sendo a associação mais forte com o PR (tabela 2). A 63 elementos (38 P+) foi aplicado o TesteRas e o TesteDx. O TesteRas foi positivo em 38 de 38 portadores com 8 falsos positivos em 27 não portadores: sensibilidade=100% e especificidade=66,67%. Na curva Roc (figura 5) para o TesteRas, o cut-off PS=252,5 tem sensibilidade=100% e especificidade=76% e o cut-off PS=260 tem sensibilidade=94,7% e especificidade=84%. O TesteDx foi positivo em 36 de 38 P+ com 3 falsos positivos: sensibilidade=94,74% e especificidade=88,89%. Por modelo logístico multivariado identifica-se um cut-off através da curva Roc que dá sensibilidade=92% e especificidade=92%. A área sob esta curva ROC é 0,927 (p <0,001), que se traduz numa capacidade admirável deste teste para discriminar entre portadores e não portadores da mutação21. A sensibilidade e especificidade de diagnóstico do ECG espontâneo usando o padrão de repolarização típico no mesmo grupo de doentes foram de 52,4 e 97,2%, respetivamente (a diferença de sensibilidade entre o TDx e o padrão de repolarização no ECG é estatisticamente significativa).
Características clínicas, demográficas e elétricas
| Portadores | Não portadores | Valor p | Associação com SCN5A (coef Eta) | |
| Características | ||||
| Demográficas e clínicas | ||||
| Sexo masculino – n (%) | 20 (47,6) | 34 (47,9) | 0,567 | - |
| Idade (anos) | 40,31±14,81 | 35,08±13,05 | 0,059 | - |
| ECG espontâneo tipo 1# – n (%) | 22 (52,4) | 2 (2,8) | <0,001 | - |
| ECG espontâneo tipo 2/3* – n (%) | 5 (11,9) | 4 (5,6) | 0,490 | - |
| Teste provocação – n total/ n positivo | 17/8 | 10/2 | 0,161 | - |
| Sintomas – n (%) | 6 (14,3) | 4 (5,6) | 0,112 | - |
| EEF – n total/ n positivo | 18/3 | 5/0 | 0,461 | - |
| Tratamento com CDI – n (%) | 8 (25,8) | - | - | - |
| ECG e ECGAR | ||||
| PR | 190,98±27,09 | 151,37±22,18 | <0,001 | 0,653 |
| dQT | 47,14±23,51 | 28,95±18,04 | <0,001 | 0,472 |
| QRSf | 106,13±9,40 | 92,41±10,76 | <0,001 | 0,544 |
| LAS | 41,92±9,75 | 31,59±11,82 | <0,001 | 0,401 |
| RMS40 | 18±6,55 | 35,26±21,8 | <0,001 | 0,47 |
Em 2008 foi demonstrado que os portadores de mutação truncante (non-sense) do SCN5A apresentam formas mais sintomáticas da doença. Foram também já descritas as perturbações da condução aurículoventricular, tanto pelo aumento do intervalo PR22,23 como do intervalo HV medido de forma invasiva por EEF na SB24. Simultaneamente, o ECGAR tem sido proposto como uma possível ferramenta para estratificação do risco arrítmico na SB25.
Contudo, a proposta de incorporação de parâmetros da condução para o seu diagnóstico é inédita com este trabalho. Desta forma, e à semelhança do que existe para a Síndrome do QT longo26 ou na cardiomiopatia arritmogénica do ventrículo direito27, o teste proposto poderia ser incorporado num score de probabilidade (tabela 3), que julgamos útil face às já discutidas fragilidades dos padrões de repolarização ventricular isolados e o peso clínico e social que este diagnóstico encerra.
Critérios diagnósticos da SB
| Critério | Pontos |
| ECG | |
| ECG tipo 1 | 5 |
| ECG tipo 2/3 | 4 |
| PS≥250 Ù dQT > 10 Ù LAS≥26 Ù RMS40≤29 | 2 |
| ECG tipo1 em elementos da família | 3 |
| Fibrilhação ventricular (FV) documentada | 3 |
| Taquicardia ventricular (TV) documentada | 3 |
| TV/FV por estimulação elétrica programada | 3 |
| Clínica | |
| História familiar de morte súbita cardíaca (< 45 anos) | 3 |
| Síncope | 3 |
| Respiração noturna agónica | 3 |
| Probabilidade de SB | |
| ≥ 8 Pontos: diagnóstico confirmado | |
| 7 Pontos: diagnóstico muito provável | |
| 6 Pontos: diagnóstico possível | |
| ≤ 5 Pontos: diagnóstico pouco provável | |
Na suspeita clínica e/ou eletrocardiográfica de SB deve ser medido (idealmente com o ECGAR) o tempo de condução intra-auricular, aurículoventricular e intraventricular (nestes 3 momentos participa o mesmo potencial de ação cardíaco de resposta rápida que está disfuncional na SB). A constatação simultânea destes 3 momentos distintos da despolarização superiores a um limite «normal-alto» (neste trabalho, esse limite foi 70 a 80% do valor teórico máximo em saudáveis) pode ter peso relativamente à probabilidade de ter ou não a doença causada por mutação do SCN5A non-sense ou missense com rebate funcional importante. Sugerimos a sua utilização na abordagem diagnóstica a familiares diretos adultos de casos índice, quando o resultado genético (ainda) não está disponível. Outra utilização possível será no cálculo de probabilidade de doença em indivíduos com ECG de Brugada idiopático ou em indivíduos com sintomas relacionados com arritmias e padrão de repolarização tipo 2 ou 3 de Brugada. Nestes casos, a informação retirada da fórmula proposta pode contribuir para reforçar (score 7), suspeitar (score 6) ou até considerá-lo pouco provável (score ≤5).
ConclusãoDemonstrámos que, isoladamente, o TesteRas e o TesteDx são uma ferramenta mais eficaz que o padrão de repolarização típico de Brugada para distinguir entre portadores e não portadores de mutações do SCN5A non-sense.
Para o diagnóstico da entidade, estes testes podem ser úteis na determinação da probabilidade de um determinado padrão de repolarização corresponder de facto a SB.
LimitaçõesO nosso trabalho tem duas grandes limitações:
- 1.
Um número relativamente reduzido de doentes portadores de apenas 2 mutações específicas. Para ficar comprovada a utilidade e reprodutibilidade do score proposto no universo de doentes com SB, será necessário aplicá-lo a outras famílias e a outros doentes com Brugada.
- 2.
A medição da onda P e do segmento PR não foi feita com a precisão do ECGAR, não dispondo o nosso grupo ainda dos meios técnicos para o fazer. A medição do intervalo PS ou ainda da OndaPf+QRSf (sem a interferência do nódulo AV) inteiramente pelo ECGAR poderá ser uma ferramenta ainda mais eficiente no score de probabilidade diagnóstica da SB.
Nesse sentido, lançamos o desafio a todos os clínicos que acompanham doentes com SB e que estejam interessados em associar-se a este trabalho para nos contactarem.
