




We report the case of a 35-year-old man admitted due to heart failure, who had had moderate cognitive deficit, craniofacial dysmorphism, epilepsy, panic attacks and congenital heart disease (subvalvular aortic stenosis) associated with chronic atrial fibrillation since childhood.
In view of his facial dysmorphism and clinical presentation, karyotype analysis was performed and revealed a de novo interstitial deletion in chromosome 8 in the region p23.1–p23.2.
This is a rare chromosomal anomaly (about 50 descriptions in the literature), whose most common manifestations include heart defects, cognitive retardation and behavioral disturbances. In this paper we present the first case with associated subvalvular aortic stenosis and review the literature on this chromosomal abnormality.
Relata-se o caso de um homem, 35 anos, internado por insuficiência cardíaca, que desde a infância apresenta quadro de défice cognitivo moderado, dismorfias faciais, epilepsia, crises de pânico e cardiopatia (estenose aórtica subvalvular) associada a fibrilhação auricular crónica.
Face às dismorfias e espectro clínico realizou cariotipo que revelou a deleção da extremidade distal do braço curto do cromossoma oito (8p23). Esta anomalia cromossómica é rara (cerca de 50 descrições na literatura), e as suas manifestações mais comuns incluem malformações cardíacas, atraso cognitivo e distúrbio comportamental. No presente artigo descreve-se o primeiro caso associado a estenose aórtica subvalvular e revê-se a literatura disponível acerca desta anomalia cromossómica.
Congenital heart defects are associated with submicroscopic chromosomal abnormalities in around 17% of cases.1 Improvements in medical and surgical treatments mean that 85% of patients with congenital heart disease now survive to adulthood.2
Deletion of the distal portion of the short arm of chromosome 8 (8p23) is a rare abnormality, whose most common manifestations include heart defects (most frequently atrioventricular septal defects), cognitive retardation and behavioral disturbances.3 There has been no report to date of association with subvalvular aortic stenosis.
Subvalvular aortic stenosis can be caused by a fibrous membrane in the left ventricular outflow tract, muscular constriction due to thickening of the subvalvular ventricular septum, or both.4 The literature contains widely varying figures for its association with other cardiovascular malformations, ranging from 13% to 71%; the most common associations are with ventricular septal defect or bicuspid aortic valve.5
Case reportWe report the case of a 35-year-old man, Caucasian, illiterate, employed in a café, who had had moderate cognitive deficit, craniofacial dysmorphism, epilepsy, panic attacks, subvalvular aortic stenosis and chronic atrial fibrillation since childhood. He had undergone cardiac surgery twice, at the ages of 12 and 24, to implant a mechanical aortic valve prosthesis, to resect a fibromuscular subaortic ring and to correct a cleft mitral valve. He was being medicated with digoxin, furosemide, acenocoumarol, carbamazepine, sodium valproate, risperidone, alprazolam and ethyl loflazepate. He had no other medical or surgical history and did not drink alcohol or smoke.
Family history included his mother who had mild facial dysmorphism (facial paresis, possibly associated with birth trauma) and mild cognitive retardation. There was no history of consanguinity, similar phenotypes, heart disease or premature or sudden death.
Around a week before admission, he began to suffer progressively worsening dyspnea on exertion, orthopnea and paroxysmal nocturnal dyspnea associated with chest pain, cough with mucous expectoration and panic attacks of increasing intensity.
On arrival at the hospital he was agitated and polypneic (respiratory rate 32 cpm), with blood pressure 111/75 mmHg, pulse 103 bpm (arrhythmic) and tympanic temperature of 36.7°C. Cardiopulmonary auscultation revealed crackling rales in the lower third of both hemithoraxes, generalized wheezing, and a metallic second heart sound, due to the mechanical valve, audible throughout the precordium. No jugular distension or peripheral edema was observed. His face (Figure 1) exhibited dolicocephaly, micrognathia, flattened base of nose, low-set ears and a wide, short neck.

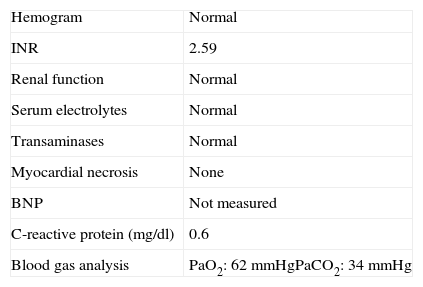
Initial diagnostic assessment revealed partial respiratory insufficiency (Table 1); the electrocardiogram (Figure 2) showed atrial fibrillation with controlled ventricular rate (90 bpm) and nonspecific ventricular repolarization abnormalities, while the posterior–anterior chest X-ray (Figure 3) revealed cardiomegaly and interstitial infiltrate in the lower third of both lung fields, suggestive of edema.
Initial diagnostic assessment.
| Hemogram | Normal |
| INR | 2.59 |
| Renal function | Normal |
| Serum electrolytes | Normal |
| Transaminases | Normal |
| Myocardial necrosis | None |
| BNP | Not measured |
| C-reactive protein (mg/dl) | 0.6 |
| Blood gas analysis | PaO2: 62 mmHgPaCO2: 34 mmHg |
BNP: brain natriuretic peptides; INR: international normalized ratio.


On echocardiography (Figure 4) there was slight left ventricular dilatation, although with good systolic function, left ventricular hypertrophy and right atrial dilatation; no signs of prosthetic valve dysfunction were observed. Pulmonary artery systolic pressure was 35 mmHg, similar to previous exams.

Karyotype analysis revealed an interstitial deletion in chromosome 8 in the region p23.1–p23.2. Cytogenetic analysis of the parents by fluorescent in situ hybridization (FISH) showed normal karyotypes.
DiscussionThe 8p23 deletion (of the most distal portion of the short arm of chromosome 8) was first described in 1988 by Fagan et al., who located the factor VII regulator on band 8p23.1.6 Since then, around 50 cases of this isolated chromosome abnormality have been reported, with no differences in gender or ethnicity. The lack of a specific phenotype, and the possibility of extremely small deletions, mean that its true prevalence is probably underestimated.8,9
Most 8p23 deletions are terminal, i.e. they include the end of the chromosome, but in some cases (including the patient presented here) interstitial deletions are found between bands 23.1 and 23.2.3 The quantity of genetic material lost in such cases may be small, and thus diagnosis requires more sensitive techniques, such as FISH.7,8 Most are de novo mutations.8
The clinical spectrum of this syndrome is highly variable, from cases so mild that the mutation was only identified in parents after it was diagnosed in their children,8,9 to the opposite extreme of mental retardation, severe heart disease and neonatal death.
The clinical manifestations include psychomotor retardation, behavioral disturbances, epilepsy, facial dysmorphism, cardiac malformations and congenital diaphragmatic hernia. Half of newborns with the syndrome have low weight, delayed development and feeding problems, and occasionally gastroesophageal reflux.10,11 After these initial difficulties, most children achieve normal height-weight development, although a degree of retardation remains in both gross8 (such as sitting and walking) and fine motor skills (such as writing).3,12
There is usually mild to moderate cognitive deficit, although with considerable interindividual variation. Language is acquired late due to difficulties with articulating various sounds, due in part to palatal abnormalities, while learning difficulties arise from attention deficit and impaired fine motor skills.3
In general these patients show normal social and affective behaviors, but there can be sudden mood changes with hyperactivity, impulsiveness and aggressiveness that may require medication.8 These changes tend to improve over time.7
Facial dysmorphism is variable, more pronounced with more extensive deletions, and can include microcephaly, dolicocephaly, flattened base of nose, short neck, low-set ears and epicanthal folds (most of which were found in the patient presented). A minority of carriers have epilepsy, mainly manifesting as absence seizures,3,7,11 although tonic–clonic seizures have been reported.8
Cardiac malformations, found in around 70% of patients, are the most important prognostic factor. Surgical correction is necessary in 40% of cases.3 A considerable variety of cardiac anomalies have been documented, of which atrioventricular septal defect is the most common.3,9,13 Other defects include pulmonary stenosis, interatrial and interventricular communications, persistent left superior vena cava, and left or right ventricular hypoplasia.14 Very rare examples have been reported of Ebstein's anomaly (two cases) and tetralogy of Fallot (three cases).3,11,14 Our literature search revealed no cases of subvalvular aortic stenosis, which makes our patient even more unusual.
Congenital diaphragmatic hernia, which hinders the normal development of the heart and lungs, is diagnosed in around 30% of patients and requires surgical correction in most cases.13,15
We found only five reports in the literature of this syndrome being diagnosed in adulthood (between 22 and 38 years of age), all with considerably more benign phenotypes than in our patient.8,9,16
The clinical variability and benign nature of some cases distinguish the 8p23 deletion from the well-characterized 8p- syndrome.8 The wide clinical spectrum of this syndrome has stimulated research into the genes responsible for its clinical manifestations, and there is evidence of a direct correlation between the size of the deletion and disease severity; deletions closer to band 23.2, or more terminal deletions, appear to lead to less severe phenotypes.9–11,17
Certain genes have been implicated in specific clinical characteristics, of which the most thoroughly investigated is GATA-4, located in the 8p23.1 region and known to be important in cardiac development through regulating genes critical to myocardial differentiation, such as those coding for troponin C and the myosin heavy chain.3,9–11,16,18,19 However, there have been reports of cases in which this gene was deleted but no cardiac malformation was found, and conversely, cases of cardiac malformation with the gene intact.9,14 Deletion of the CGAT (chromaffin granule amine transporter) and TNKS (tankyrase 1) genes has been implicated in behavioral disturbances and learning difficulties due to the high level of their expression in the brain9; the latter has also been linked to diaphragmatic hernias. Regions of chromosome 8 associated with microcephaly and facial dysmorphism have also been investigated.3,10,15
To summarize, in the case presented we highlight a rare chromosomal abnormality associated with heart disease that can be surgically corrected and that has a wide clinical spectrum, in which each new case reported can help complete the picture of the alterations associated with the syndrome.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data and that all the patients included in the study received sufficient information and gave their written informed consent to participate in the study.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
Conflicts of interestThe authors have no conflicts of interest to declare.
We thank Dr. Ana Madeira of the Genetics Department of Centro Hospitalar Lisboa Norte, EPE.
Please cite this article as: Aguiar, P. Estenose aórtica subvalvular associada à deleção 8p23. Rev Port Cardiol. 2013. http://dx.doi.org/10.1016/j.repc.2012.05.025.
