




Cardiovascular mortality has fallen in Portugal over the last decade, but mortality from stroke has declined much more sharply than from ischemic heart disease. Data from the European Commission show that from 1998–2000 to 2007–2009 mortality from stroke fell from 145.2 to 78.5 per 100000 population, while death from ischemic heart disease only decreased from 65.7 to 44.6 per 100000 population (age-adjusted rates).
According to the Portuguese National Coordinating Body for Cardiovascular Disease, of the 18075 patients admitted to cardiac care units during 2010, 5320 were admitted for non-ST elevation myocardial infarction (MI) and 4308 for ST-elevation MI, an increase of 7.8% and 8.6%, respectively, compared to 2009. Primary percutaneous coronary intervention (PCI) was the method of reperfusion in 2838 patients, 10.9% more than in the previous year. In 2009, there were 12318 PCI procedures – 6.6% more than in 2009 – in which 15122 stents were implanted, 10590 of them drug-eluting.
Acute coronary syndromes (ACS) require intensive treatment in the acute phase to alleviate myocardial ischemia and to prevent adverse events such as death, (re)infarction and potentially fatal arrhythmias. After clinical stabilization and hospital discharge, these patients still have relatively high rates of thrombotic complications, and thus intensive therapy needs to be maintained in the chronic phase. Recurrence of ACS, which can be fatal, in the weeks or months following discharge can have at least two causes. Firstly, intracoronary ultrasound studies show that most patients with ACS have at least two simultaneous plaque ruptures, and some have ruptured plaques in all three coronary arteries.1 Consequently, a focal therapeutic approach aimed at treating the culprit lesion, which can often be achieved by PCI, does not remove the risk of short-term recurrence of instability due to other highly vulnerable plaques. Secondly, ACS are characterized by inflammation and oxidative stress.2 In inflammatory states, circulating monocytes produce thromboxane A2 from arachidonic acid via the cyclooxygenase-2 pathway, an extra-platelet source of thromboxane A2 that is not inhibited by low doses of aspirin. In an oxidative environment, prostanoids are produced by non-enzymatic peroxidation from arachidonic acid in the cell membrane and circulating low-density lipoproteins. These increase platelet reactivity, leading to activation even in the presence of subthreshold concentrations of agonists.
Platelets play a crucial role in the pathogenesis of ACS and their complications, and a combination of antiplatelet agents with different therapeutic targets strengthens platelet inhibition.3 An important step in platelet activation is the release of granules containing adenosine phosphate (ADP), which binds to both the P2Y1 and P2Y12 receptors. Unlike the P2Y1 receptor, the P2Y12 receptor is found in few tissues and is thus a more promising therapeutic target. It is the binding of ADP to the P2Y12 receptor that increases platelet activation; inhibiting these receptors makes the thienopyridines more potent antiplatelet agents than aspirin, since ADP enables platelet aggregation even with low concentrations of any platelet agonists.
In the light of these mechanisms, studies have demonstrated the superiority of dual platelet inhibition with aspirin and clopidogrel in non-ST elevation ACS as well as in ST-elevation MI.4,5 The benefit of dual antiplatelet therapy is apparent after only 24hours of treatment and is independent of baseline clinical characteristics and therapeutic approach (with or without PCI). The European Society of Cardiology accordingly recommends dual antiplatelet therapy with aspirin and a P2Y12 receptor antagonist for all ACS patients, to be initiated as soon as possible and to be maintained for a year if bleeding risk permits.
Despite the considerable improvements due to dual antiplatelet therapy with aspirin and clopidogrel, significant thrombotic risk remains in both the short and long term. The existence of marked interindividual variability in response to clopidogrel indicates that this residual risk is modifiable to some extent.6 A weak response to clopidogrel is clinically relevant, since it is associated with increased risk of cardiovascular death, spontaneous MI, type 4a MI, stent thrombosis, stroke and target lesion revascularization,7 and it is common (around a third of patients), because the ratio between the recommended clopidogrel dose and the minimum dose required to obtain its maximum pharmacodynamic effect is around one, which means that the usual dose only partially inhibits P2Y12 receptors.8
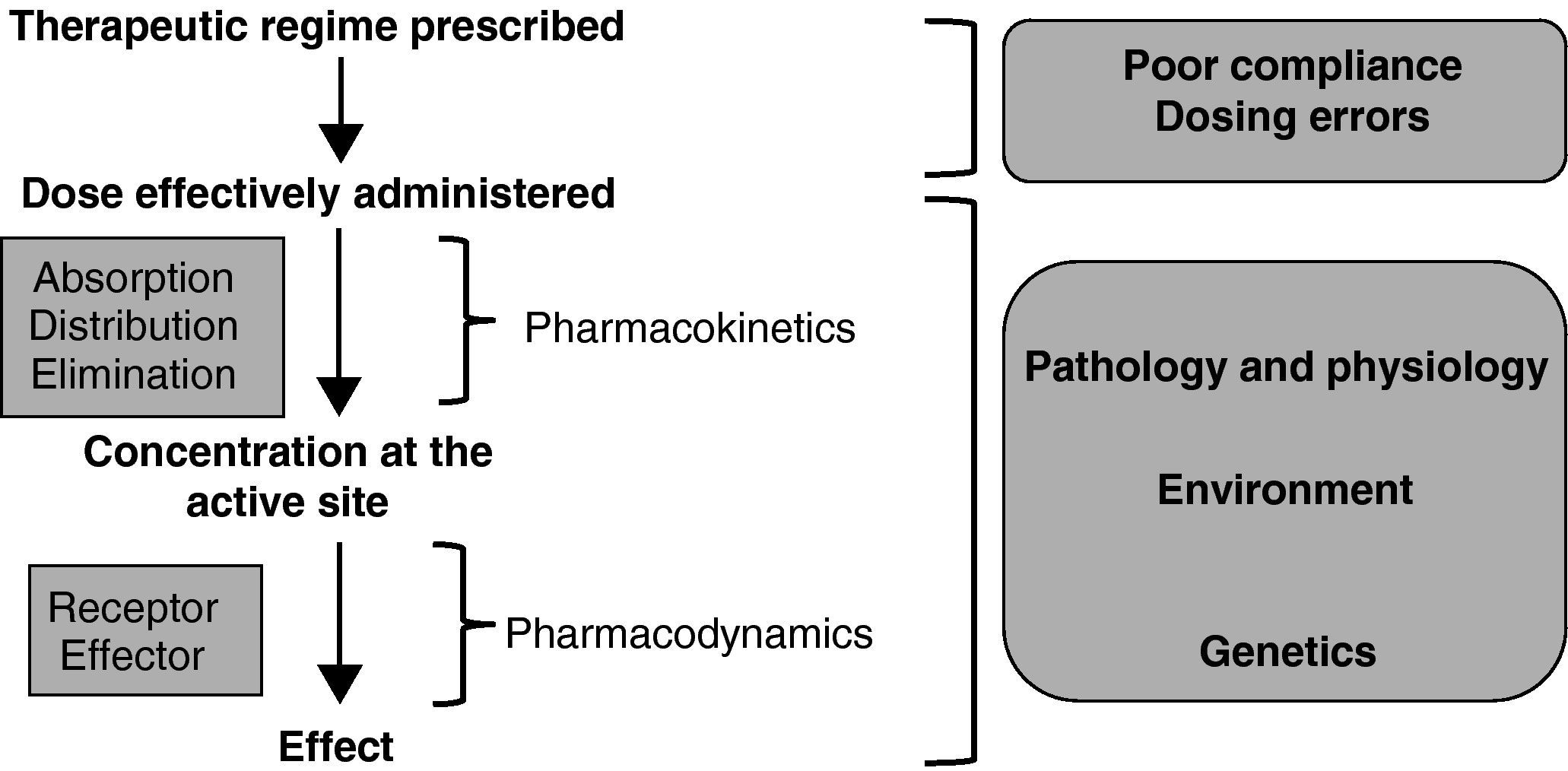
Figure 1 shows the sequence of events required to achieve the desired therapeutic effect of any drug.
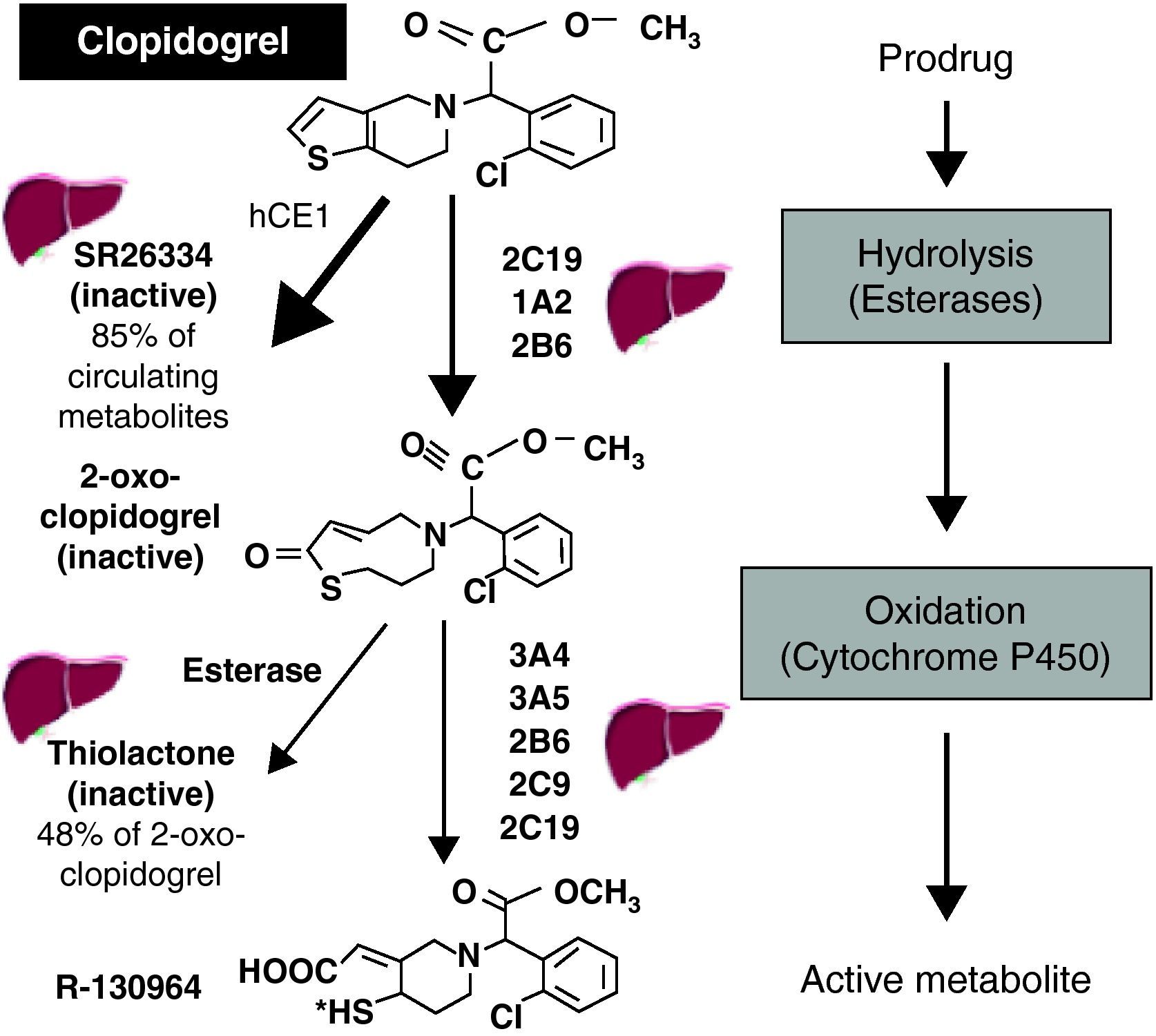
Many factors affect the response to clopidogrel, some of which have been the subject of considerable research, particularly polymorphisms in the ABCB1 gene, which codes for P-glycoprotein (an efflux pump in the intestinal epithelium that returns absorbed drugs to the intestinal lumen, thereby reducing their bioavailability), and in the gene that codes for the 2C19 isoenzyme of cytochrome P450 (CYP2C19), which is important for the activation of clopidogrel.9 Clopidogrel is a prodrug, and its activation is complex (Figure 2).7 Some 85% of the dose administered is hydrolyzed in the liver, where it is converted into an inactive metabolite (SR 26334); the remainder is metabolized by CYP2C19 and to a lesser degree by CYP1A2 and CYP2B6 to 2-oxo-clopidogrel. Around half of this metabolite, which is also inactive, is hydrolyzed and converted into an inactive thiolactone, while the other half is metabolized by the 3A4, 3A5, 2B6, 2C9 and 2C19 isoenzymes, finally producing the active metabolite (R-130964). The fact that a significant proportion of the absorbed drug is wasted (converted to inactive metabolites), the complex activation of the prodrug requiring two oxidation steps, and its heavy dependence on the function of the CYP2C19 isoenzyme (which is involved in the metabolization of numerous drugs and whose reduced-function genetic variants have a prevalence of around 25%), are the main pharmacokinetic factors affecting the variability of response to clopidogrel.
The article by Teixeira et al. published in this issue of the Journal confirms the prognostic impact of reduced-function CYP2C19 variants in a Portuguese population.10 The authors found these variants in 27% of a sample of 95 patients who survived ACS and were medicated with clopidogrel. Despite the study's limitations (particularly the small sample size), its main findings are in agreement with those of several other analyses. Recent studies suggest that reduced-function CYP2C19 variants are clinically relevant only in patients undergoing PCI with stenting, in whom they are associated with greater risk for MI and stent thrombosis.11
The question thus arises as to how to optimize platelet inhibition in ACS. Should platelet response to clopidogrel be tested? Should genetic variants that affect clopidogrel response be screened for? What should be done in the presence of weak platelet inhibition or a reduced-function genetic variant? Would it be better to use one of the new P2Y12 receptor antagonists, which provide faster, stronger and more consistent platelet inhibition? To put it another way, do we need personalized antiplatelet therapy or better antiplatelet agents?
The concept of personalized medicine has significant limitations. It requires laboratory tests, but the ideal tests have not been identified, and existing tests lack standardization and clinical validation. Furthermore, a personalized approach to platelet therapy is a lengthy and costly process, and will never be 100% effective, firstly because some patients, when tested for response to clopidogrel, still present high platelet reactivity even after a 2400-mg loading dose and 300-mg maintenance dose,12,13 and secondly, because genetic variants in CYP2C19 explain only 12% of the variability of response to clopidogrel.14 In the study by Teixeira et al., this may have contributed to the lack of impact of genotype on the results of platelet function tests.
The future undoubtedly lies with the new P2Y12 receptor antagonists, particularly prasugrel and ticagrelor. These drugs are more effective than clopidogrel, even when administered in doses above those recommended.15 In the TRITON-TIMI 38 trial, in individuals with ACS with scheduled PCI, prasugrel reduced major vascular events by 19%, but increased the risk of major bleeding; the risk/benefit ratio was better than for clopidogrel except in patients with a history of stroke or transient ischemic attack.16 In the PLATO trial, ticagrelor reduced the incidence of major vascular events in patients with scheduled primary PCI or non-ST elevation ACS by 16%; it increased major bleeding risk compared to clopidogrel but did not increase risk for CABG-related bleeding.17 It is hoped that these new drugs will reduce the high thrombotic risk associated with ACS and contribute significantly to further decreases in mortality from ischemic heart disease.
Conflicts of interestThe author has no conflicts of interest to declare.
Please cite this article as: Inibição Plaquetária nas Síndromes Coronárias Agudas: Uma Intervenção Complexa que Deve Ser Optimizada. Rev Port Cardiol 2012. doi:10.1016/j.repc.2012.02.011.
