




Holt-Oram syndrome is clinically characterized by morphological abnormalities of the upper limbs and congenital cardiac defects. Although the disease is congenital, the diagnosis may only be made later in life. It is a rare autosomal dominant disorder, caused by a mutation in the TBX5 gene located on chromosome 12, but sporadic cases have also been reported. We describe the case of a 75-year-old man with known morphological alterations of the upper limbs since birth and congenital cardiac defect (atrial septal defect), who later in life also manifested with advanced atrioventricular block.
A síndrome de Holt-Oram é caracterizada pela associação de defeitos morfológicos dos membros superiores com defeitos cardíacos congénitos. Apesar de ser uma doença congénita, o seu diagnóstico pode só ser feito numa idade mais avançada. É uma doença autossómica dominante rara, devido à mutação do gene T-BX5, localizado no cromossoma 12, mas casos esporádicos também já foram reportados. Descrevemos o caso de um doente de 75 anos, com alterações morfológicas dos membros superiores, presentes desde o nascimento e defeito cardíaco (comunicação inter-auricular) diagnosticado na adolescência, que numa idade mais avançada apresentou bloqueio aurículo-ventricular completo.
Holt-Oram syndrome (HOS), also known as hand-heart syndrome, is a rare genetic disorder clinically characterized by morphological abnormalities of the upper limbs and congenital cardiac defects. Several mutations have been described, but the most frequent is in the TBX5 gene of the T-box complex, located on chromosome 12. Upper limb abnormalities are always present, and the presence of lower limb abnormalities excludes the diagnosis. Several heart malformations have been described, but atrial septal defect and ventricular septal defect are the most common. Since clinical manifestations may be subtle, the diagnosis may only be made later in life, or even missed.
Case reportWe present the case of a 75-year-old man with a previous history of morphological defects of both upper limbs since birth, although no etiological study was ever made. At the age of 15, in the context of tiredness, he was assessed by a cardiologist and an ostium secundum atrial septum defect (ASD) was diagnosed. The patient reported having a low heart rate, around 40 bpm, from a young age, but was always asymptomatic, and so a 24-hour Holter recording was performed annually. From the age of 58 he had documented atrial fibrillation.
At the age of 62, he underwent surgical repair of the ASD, with a continuous suture, in the context of increasing fatigue and dyspnea on exertion and recurrent respiratory infections.
After this surgery he remained asymptomatic from the cardiovascular standpoint, reporting no loss of consciousness, tiredness or breathlessness.
Adding to the described cardiac history, at the age of 55 the patient was diagnosed with Ménière's syndrome, and suffered occasional periods of exacerbation with vertigo, which improved with betahistine treatment.
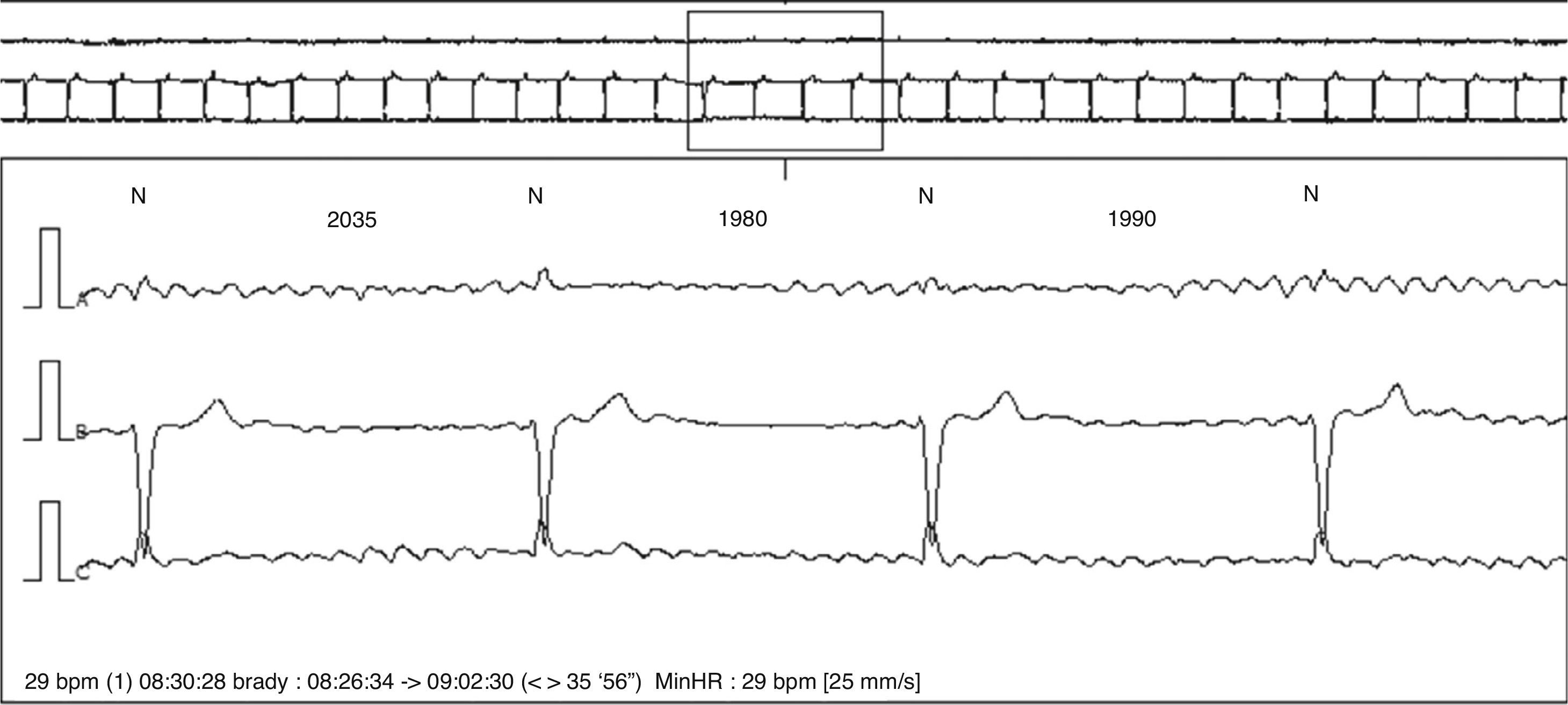
At the age of 75, he began to have symptoms of dizziness and tiredness, but denied other symptoms, including loss of consciousness, palpitations, dyspnea or chest pain. He was medicated with betahistine, and a 24-hour Holter recording was ordered by his general practitioner. This showed atrial fibrillation as base rhythm, with marked bradycardia (25 bpm); 8909 pauses were registered, predominantly nocturnal, the longest lasting 2.76 seconds in the context of complete atrioventricular block (Figure 1); throughout the recording the patient remained asymptomatic. He was referred for an outpatient cardiology consultation for clinical assessment and treatment in our institution.
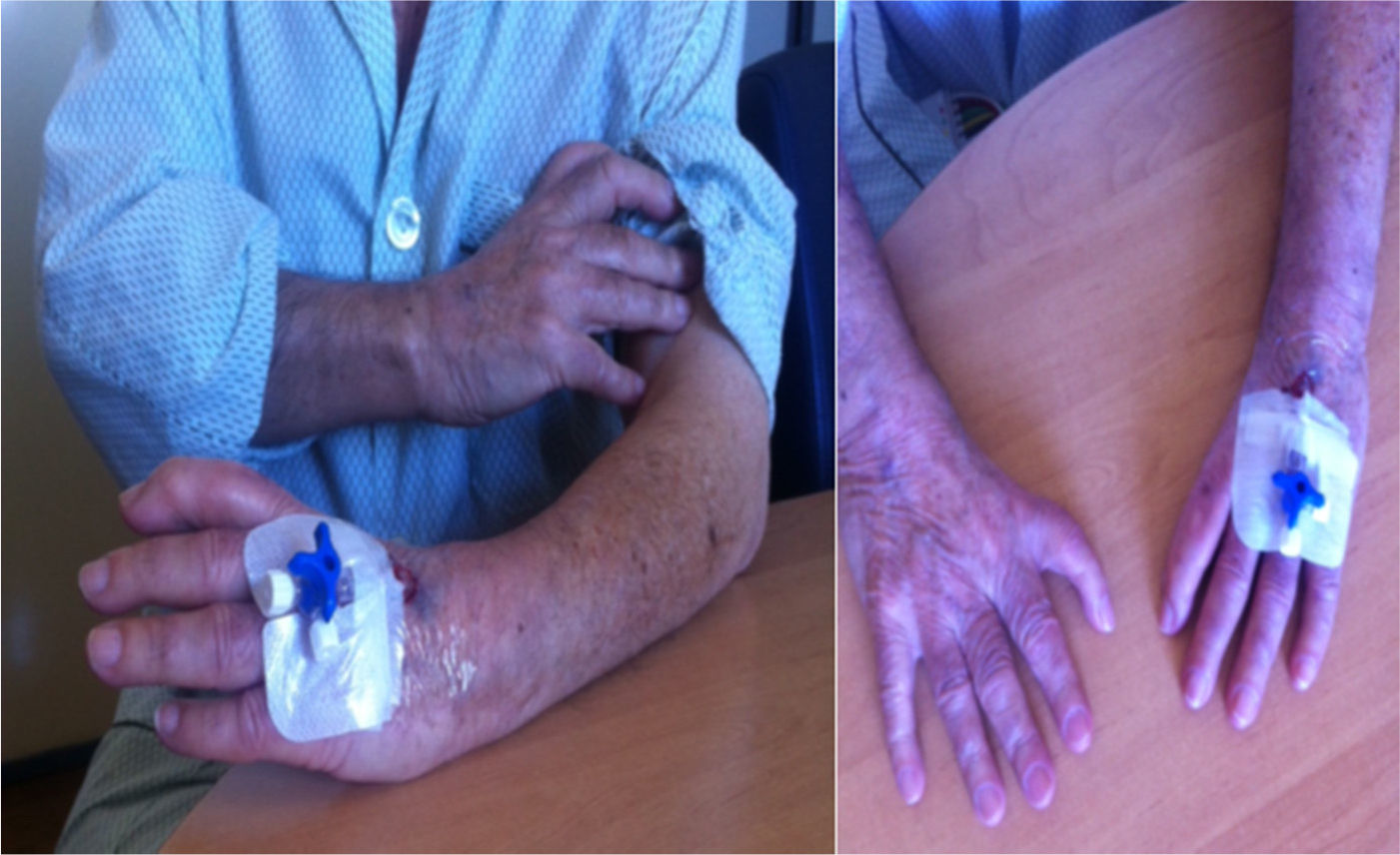
After cardiac assessment and additional medical tests, a permanent VVI pacemaker was implanted due to complete atrioventricular block in a patient with atrial fibrillation. During this hospitalization, the physical examination was notable for asymmetry of the upper limbs; morphological changes in the first finger of the right hand, showing similar morphology to the other fingers, and absence of one finger of the left hand (Figure 2); and a central linear scar in the sternum, related to the previous cardiac surgery. No other significant changes were found on physical examination.
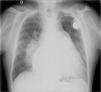
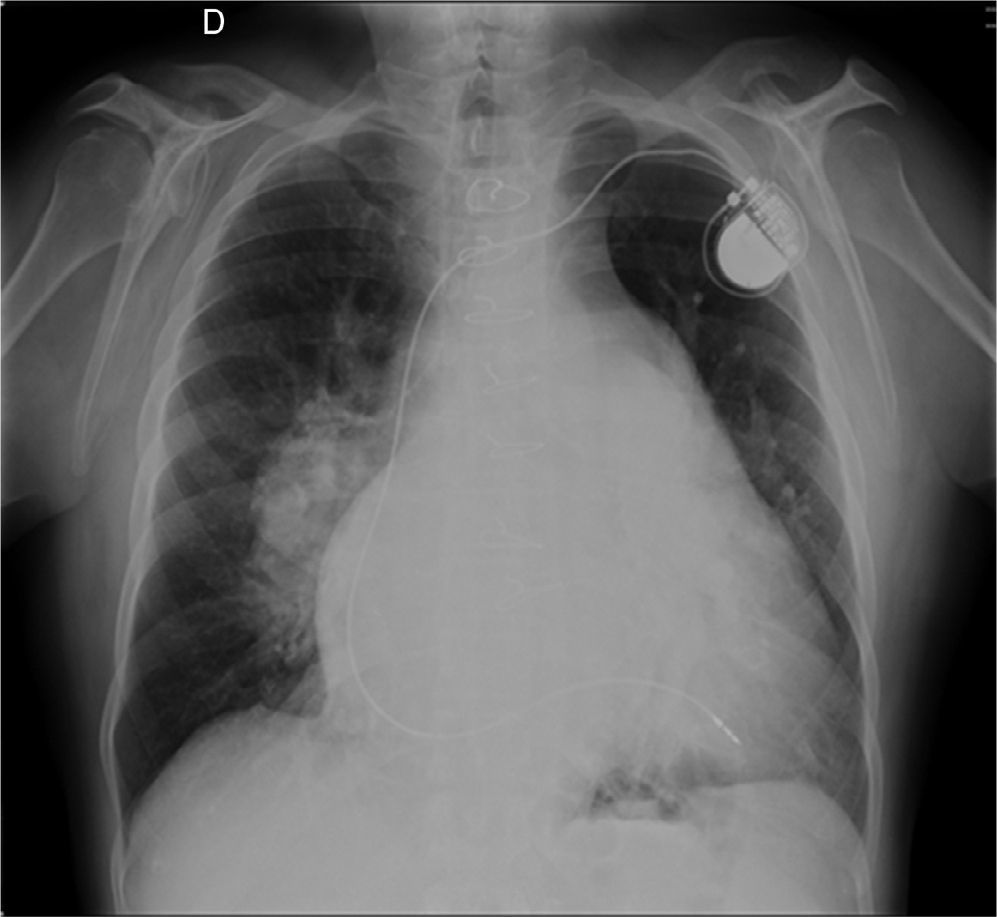
The electrocardiogram showed atrial fibrillation with ventricular response of 55 bpm, right bundle branch block and left posterior hemiblock. The chest X-ray after pacemaker implantation (Figure 3) showed a cardiothoracic ratio of >50%, dilatation of the right atrium and of both pulmonary arteries, and pulmonary vasculature enhancement. The transthoracic echocardiogram showed a dilated left atrium; interventricular septal hypertrophy, with preserved left ventricular function, and dilated right chambers with annular dilation of the tricuspid valve, causing moderate tricuspid valve regurgitation, with pulmonary artery systolic pressure calculated at 50 mmHg (moderate pulmonary hypertension); and a dilated pulmonary artery and mild pulmonary regurgitation. No morphological defect of the atrial septum was found, or flow across the atrial septum; the ventricular septum also showed no morphological defect.
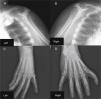
The patient also underwent X-ray of the shoulder girdle and upper arm, which showed hypoplasia of the glenoid cavity and bilateral degenerative changes of the gleno-humeral joints; bilateral X-ray of the elbow, fist and hand showed right elbow luxation in the radio-humeral joint, with normal morphology of the left elbow, morphological changes to the proximal carpal row of the right fist, the first finger of the right hand morphologically similar to the others, and syndactyly between the first and second axes of the left hand and wrist; the trapezium and trapezoid could not be differentiated (Figure 4).
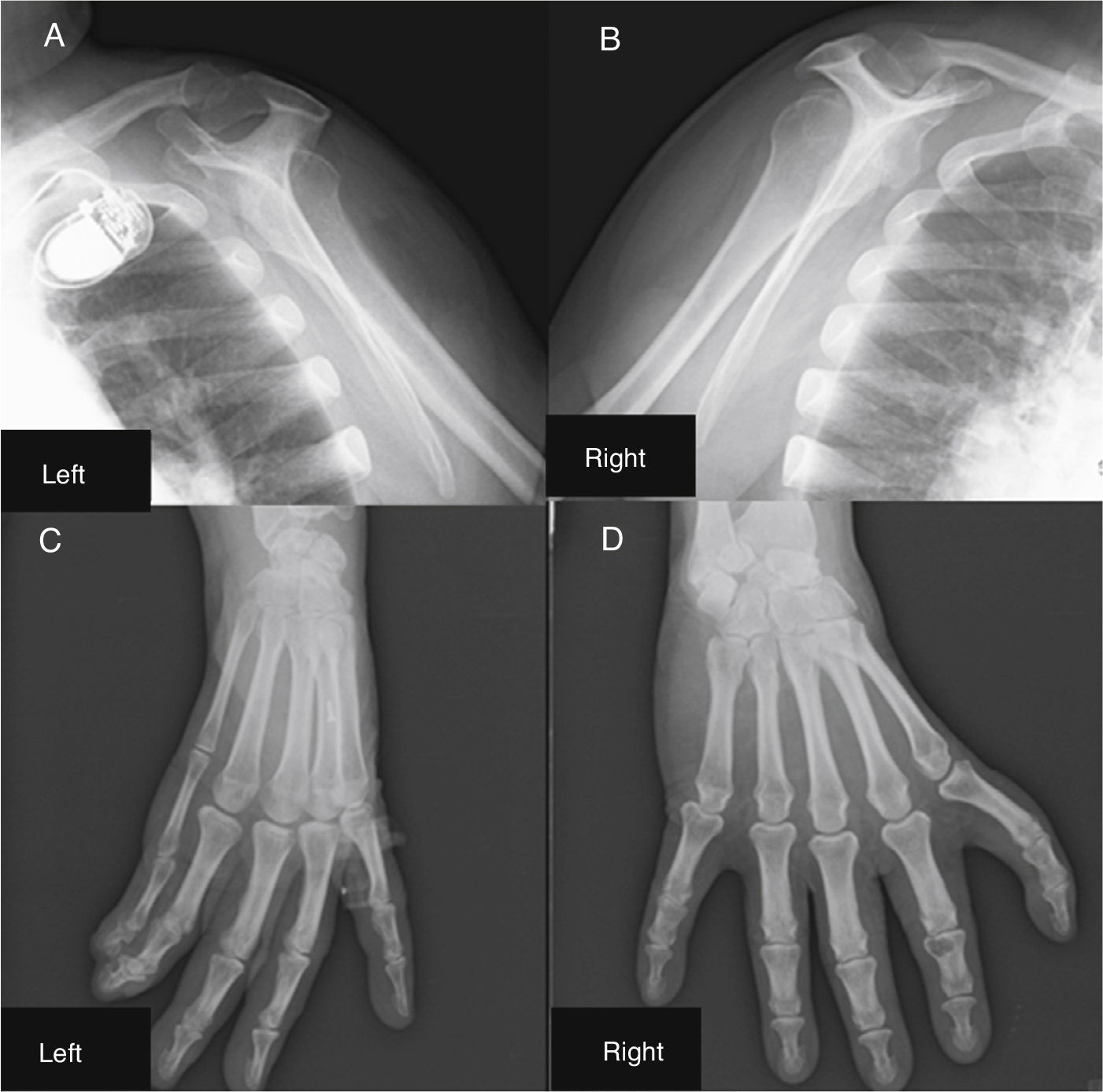
Shoulder girdle X-ray showing hypoplasia of the glenoid cavity and bilateral degenerative changes of the gleno-humeral joints (A and B); X-ray showing syndactyly between the first and second axes of the left hand and wrist; the trapezium and trapezoid cannot be differentiated (C); right hand with morphological changes of the proximal carpal row; the first finger of the right hand is morphologically similar to the others (D).
No morphological abnormalities were found on lower limb X-ray.
A genetic study was requested but the results are not yet available.
Since this admission, the patient has been followed in outpatient cardiology consultations; he has remained asymptomatic, medicated with warfarin, losartan, hydrochlorothiazide, pravastatin and betahistine.
Investigation of the patient's family history revealed that he had had a brother who died soon after birth (cause of death unknown); apparently there are no other cases of skeletal dimorphism or cardiac defects in the family.
DiscussionHOS was first described in 1960 by Mary Holt and Samuel Oram,1,2 in a four-generation family with atrial septal defects and thumb abnormalities.1–3 It is a rare disorder with a prevalence of one case per 100 000 births in the USA.4–6
It is an autosomal dominant condition4–8 with genetic heterogeneity, but the most common mutations,7 present in 70% of patients with clinical diagnosis,9 are in the TBX5 gene of the T-box complex, located on chromosome 12 (12q 24.1), which encodes a transcription factor7 that is important in the development of both the heart and upper limbs.4 Although most cases are of familial transmission, the literature describes cases due to new mutations6,8 in 40–85% of cases.
Clinical manifestations are variable, but upper limb abnormalities are always present. Defective development of the embryonic radial axis (e.g. aplasia, hypoplasia, fusion, or other anomalous development) results in a wide spectrum of phenotypes, including triphalangeal or absent thumbs, foreshortened arms and phocomelia (a malformation in which the hands are attached close to the body; also termed pseudothalidomide syndrome).5,8 Abnormalities may be unilateral or bilateral and asymmetric, and may involve the radial, carpal, and thenar bones. Left-sided hand and arm abnormalities are often more severe than right-sided ones.5
The most prevalent findings in patients with HOS are malformations or fusions of the carpal bones. Carpal bone abnormalities are the only findings present in every affected individual, although these anomalies may be evident only radiographically in some patients.5 Other radiographic abnormalities include posteriorly and laterally protuberant medial epicondyles of the humerus, hypoplastic clavicles, shortened radii, and ulnar hypoplasia (occurring only in patients with radial defects).5 Clinical recognition of subtle limb anomalies in patients with HOS may require both physical examination and radiographs of the upper extremities,5 and assessment of these patients should include lower limb X-ray, since lower limb abnormalities exclude the diagnosis. The most common alterations have been described earlier. Individuals without carpal bone abnormalities in the preaxial radial bones do not have HOS.
In the present case the patient had evident abnormalities of both upper limbs since birth that were subsequently confirmed by typical radiographic findings, with hypoplasia of the glenoid cavity, bilateral degenerative changes of the gleno-humeral joints, morphological changes to the proximal carpal row of the right hand, the first finger of the right hand morphologically similar to the others, and syndactyly between the 1st and 2nd axes of the left hand and wrist; the trapezium and trapezoid could not be differentiated.
The patient presented in this case report denies that any other family member has upper limb morphological abnormalities or known cardiac defects. This may be due to two possibilities: either family members have a subtle morphological upper limb abnormality with or without cardiac defect, or this is a case of a new mutation in the TBX5 gene.
Clinically all patients with HOS have upper limb anomalies6 and 85–95% have cardiac malformations.5 Lower limb abnormalities have not been reported.
The clinical diagnostic criteria of HOS are personal and/or family history of cardiac septation and/or conduction defects in the setting of preaxial radial ray axis deformity.9
As stated above, the majority of patients also have cardiac malformations5,8 and almost every type of cardiac anomaly has been reported, either singly or as part of a group of multiple defects.5 The most common are ASD and ventricular septal defect (VSD), which vary in number, size, and location.2,8 The most common ASD is of the secundum type2,5; VSDs are usually located in the muscular trabeculated septum.8 Rhythm abnormalities sometimes accompany the morphological abnormalities,8 including atrioventricular block with bradycardia, atrial fibrillation and sudden death from heart block.5,8
Our patient had the most common type of cardiac defect, ostium secundum type ASD, which was repaired surgically. Later in life he developed arrhythmic disorders, initially presumably sinus bradycardia, which over the years developed into atrial fibrillation and atrioventricular block, as documented on 24-hour Holter recording.
As a congenital disease, HOS is present at birth in all patients, although abnormalities may not be clinically apparent until later in life, when cardiac symptoms occur, or when a child of the family has a more severe presentation. Cardiac conduction disease tends to progress with age, middle-aged individuals often presenting with significant atrioventricular block or atrial fibrillation.10
Chest X-ray may show enlarged pulmonary arteries due to pulmonary hypertension or cardiomegaly and evidence of congestive heart failure may be present.
The echocardiogram is the procedure of choice to detect morphological abnormalities of the heart. Apart from the common ASD and VSD, other cardiac anomalies may include abnormal isomerism and anomalous pulmonary venous return.
ECG or 24-hour Holter recording may be valuable in the diagnosis and follow-up of rhythm abnormalities.
HOS is a rare disorder and little is known about its prognosis. Current evidence shows that prognosis depends on the severity of the cardiac manifestations; patients with severe morphological or electrical cardiac manifestations have worse prognosis and may need surgery, device implantation or specific therapy. In cases of mild cardiac manifestations, patients may have a close to normal life expectancy.
Although our patient had morphological limb abnormalities and cardiac manifestations (first morphological and then arrhythmic), these did not prevent him from leading a normal life.
ConclusionWe describe the case of a patient with a rare syndrome which went undiagnosed until late in life. Although he had manifestations from birth of both upper limb and cardiac disorders, a link between these two disorders was not established until later in life, when he developed third-degree atrioventricular block with need for pacemaker implantation, although he had been followed by a cardiologist and a general practitioner.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that the procedures followed were in accordance with the regulations of the relevant clinical research ethics committee and with those of the Code of Ethics of the World Medical Association (Declaration of Helsinki).
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
Conflicts of interestThe authors have no conflicts of interest to declare.
