


















The Acute Dialysis Quality Initiative consensus conference proposed a classification of cardiorenal syndrome (CRS), aiming for a better delineation of each subtype. Although the exact pathophysiology of type 4 CRS is not completely understood, the mechanisms involved are probably multifactorial. There is growing evidence that oxidative stress is a major connector in the development and progression of type 4 CRS. Giving its complexity, poor prognosis and increasing incidence, type 4 CRS is becoming a significant public health problem. Patients with chronic kidney disease are particularly predisposed to cardiac dysfunction, due to the high prevalence of traditional cardiovascular risk factors in this population, but the contribution of risk factors specific to chronic kidney disease should also be taken into account.
Much remains to be elucidated about type 4 CRS: despite progress over the last decade, there are still significant questions regarding its pathophysiology and there is as yet no specific therapy. A better understanding of the mechanisms involved may provide potential targets for intervention.
The present review will provide a brief description of the definition, epidemiology, diagnosis, prognosis, biomarkers and management strategies of type 4 CRS, and the pathophysiological mechanisms and risk factors presumably involved in its development will be particularly highlighted.
A definição e classificação de síndrome cardiorrenal foram avançadas pela conferência Acute Dialysis Quality Initiave, visando uma melhor compreensão acerca de cada subtipo desta patologia. Embora a fisiopatologia exata não esteja completamente definida, os mecanismos envolvidos na síndrome cardiorrenal tipo 4 são potencialmente multifatoriais. Há crescente evidência de que o stress oxidativo é um conector principal no desenvolvimento e progressão desta síndrome. Dada a sua complexidade, pior prognóstico e incidência crescente, a síndrome cardiorrenal tipo 4 tem vindo a tornar-se num importante problema de saúde pública. Os pacientes com doença renal crónica estão particularmente predispostos a desenvolver disfunção cardíaca, devido à elevada prevalência de fatores de risco cardiovasculares nesta população, mas a contribuição de fatores de risco específicos da doença renal crónica devem ser tidos em conta.
Muito permanece por esclarecer acerca da síndrome cardiorrenal tipo 4: apesar do progresso ao longo da última década, persistem questões importantes relativamente à sua fisiopatologia e ainda não existe tratamento específico. É sublinhada a necessidade de elaborar recomendações científicas para a prevenção e estratégias terapêuticas eficazes para a síndrome cardiorrenal tipo 4, sendo que um conhecimento mais aprofundado acerca dos mecanismos envolvidos na síndrome cardiorrenal tipo 4 poderão conferir potenciais alvos de intervenção.
A presente revisão irá focar-se na síndrome cardiorrenal tipo 4: a sua definição, epidemiologia, diagnóstico, prognóstico, biomarcadores e estratégias terapêuticas serão brevemente descritos, mas uma particular atenção será dada aos mecanismos fisiopatológicos e fatores de risco presumivelmente envolvidos nesta patologia.
coronary artery disease
chronic kidney disease
cardiorenal syndrome
ejection fraction
endoplasmic reticulum
end-stage renal disease
fibroblast growth factor 2
fibroblast growth factor 23
glomerular filtration rate
hemodialysis
heart failure
interleukin
indoxyl sulphate
left ventricular
nitric oxide
protein-bound uremic toxins
peritoneal dialysis
parathyroid hormone
renin-angiotensin-aldosterone system
reactive oxygen species
sudden cardiac death
sympathetic nervous system
transforming growth factor-beta
tumor necrosis factor-alpha
vitamin D receptor
The heart and kidneys are so closely connected that each dysfunctional organ has the ability to initiate and perpetuate injury in the other, through hemodynamic, neurohormonal and immunologic/biochemical feedback pathways.1,2 To reinforce this crosstalk between heart and kidneys, the concept of the cardiorenal syndrome (CRS) was advanced by the Acute Dialysis Quality Initiative (ADQI), defined as “a disorder of the heart and kidneys whereby acute or chronic dysfunction in one organ may induce acute or chronic dysfunction of the other”.3–9 It has been divided into five subtypes (Table 1), according to the primary organ dysfunction and time frame, as well as the clinical context.5,8–10 This classification is not intended to be static: many patients may transition between different CRS subtypes during the course of their disease.10–13 The interplay between cardiac and renal disease is complex, with varying patterns over time and a variable degree of reversibility.14
Classification of cardiorenal syndrome subtypes.
| Description | |
|---|---|
| Type 1 | Abrupt worsening of cardiac function (e.g. acute cardiogenic shock or acute decompensated HF) leading to AKI |
| Type 2 | Chronic abnormalities in cardiac function (e.g. chronic congestive HF) causing progressive and permanent CKD |
| Type 3 | Abrupt worsening of renal function (e.g. AKI) causing acute cardiac disorder (e.g. acute HF) |
| Type 4 | CKD contributing to decreased cardiac function, cardiac hypertrophy, fibrosis and/or increased risk of adverse cardiovascular events |
| Type 5 | Systemic condition (e.g. sepsis) causing both acute cardiac and renal injury and dysfunction |
CRS is a common condition, as cardiac and renal diseases and their risk factors become increasingly prevalent, and is associated with poor clinical outcomes. Patients who develop CRS have an increased risk of hospital admission and high mortality: when kidney and heart failure coexist, there is progressive dysfunction of each organ and survival is negatively affected.15–17
The present review will focus on type 4 CRS. Firstly, the characteristics of chronic kidney disease (CKD) and heart failure (HF) will be briefly described, since these conditions are key concepts in the definition of type 4 CRS. Then, the definition, epidemiology, diagnosis, prognosis, biomarkers and treatment of type 4 CRS will be summarized. Finally, particular attention will be paid to the pathophysiology of type 4 CRS and the risk factors involved in its development and progression.
Chronic kidney diseaseDefined as abnormalities of kidney structure or function or reduced renal function (glomerular filtration rate [GFR] <60 ml/min/1.73 m2) present for at least three months, CKD is characterized by progressive destruction of kidney parenchyma due to fibrosis (Table 2).18–22 Kidney damage is defined as pathological abnormalities or markers of damage, including abnormalities in blood or urine tests or imaging studies.23 CKD can be categorized according to GFR and albuminuria categories (Table 3).22
Definition of chronic kidney disease.
| Criteria for CKD (either of the following present for more than three months) | |
|---|---|
| Markers of kidney damage (one or more) | Albuminuria Urine sediment abnormalities Electrolyte and other abnormalities due to tubular disorders Abnormalities detected by histology Structural abnormalities detected by imaging History of kidney transplantation |
| Decreased GFR | GFR <60 ml/min/1.73 m2 (GFR categories G3a-G5) |
CKD: chronic kidney disease; GFR: glomerular filtration rate.
Adapted from KDIGO 2012.22
Glomerular filtration rate and albuminuria categories in chronic kidney disease.
| GFR category | Terms | GFR (ml/min/173 m2) |
|---|---|---|
| G1 | Normal or high | ≥90 |
| G2 | Mildly decreased | 60-89 |
| G3a | Mildly to moderately decreased | 45-59 |
| G3b | Moderately to severely decreased | 30-44 |
| G4 | Severely decreased | 15-29 |
| G5 | Kidney failure, including ESRD | <15 |
| Albuminuria category | Terms | AER (mg/24 h) | ACR (mg/g) |
|---|---|---|---|
| A1 | Normal to mildly increased | <30 | <30 |
| A2 | Moderately increased | 30-300 | 30-300 |
| A3 | Severely increased | >300 | >300 |
In the absence of evidence of kidney damage, neither GFR category G1 nor G2 fulfill the criteria for CKD.
ACR: albumin-to-creatinine ratio; AER: albumin excretion rate; CKD: chronic kidney disease; ESRD: end-stage renal disease; GFR: glomerular filtration rate.
Adapted from KDIGO 2012.22
The worldwide prevalence of CKD is 8-16% and increasing, and it is becoming a significant health problem with high morbidity, mortality and treatment costs.20 Patients with CKD may need renal replacement therapy or renal transplant in the future.18
CKD is a well-known independent cardiovascular risk factor.24 Cardiovascular risk increases with decreasing renal function: even mild decreases are associated with significantly worse cardiovascular outcomes and mortality (Figure 1).5,8,19,25–29 CKD is associated with various complications, morbidity and mortality (Table 4).22 CKD patients have a 10- to 30-fold increased risk for cardiac mortality compared to age- and gender-matched controls.5,6,25–28,30 Thus, CKD acts as a risk multiplier, accelerating the development of cardiovascular disease and increasing the risk of death.5,19 In addition, the spectrum of cardiovascular disease in advanced stages of CKD appears to have its own unique characteristics: while coronary artery disease (CAD) is the major cardiovascular cause of death in the general population, primary cardiac arrhythmias account for 50% of cardiovascular deaths in patients with end-stage renal disease (ESRD).30–32
 GFR among 1120295 ambulatory adults. Cardiovascular events included hospitalization for coronary artery disease, heart failure, stroke or transient ischemic attack, and peripheral arterial disease.
GFR among 1120295 ambulatory adults. Cardiovascular events included hospitalization for coronary artery disease, heart failure, stroke or transient ischemic attack, and peripheral arterial disease. Age-standardized rates of death from any cause, cardiovascular events and hospitalization, according to estimated GFR among 1120295 ambulatory adults. Cardiovascular events included hospitalization for coronary artery disease, heart failure, stroke or transient ischemic attack, and peripheral arterial disease. GFR: glomerular filtration rate.
Prevalence of chronic kidney disease complications by glomerular filtration rate category.
| Complication | GFR category (ml/min/1.73 m2) | ||||
|---|---|---|---|---|---|
| ≥90 | 60-89 | 45-59 | 30-44 | <30 | |
| Anemia | 4.0% | 4.7% | 12.3% | 22.7% | 51.5% |
| Hypertension | 18.3% | 41.0% | 71.8% | 78.3% | 82.1% |
| 25-OH vitamin D deficiency | 14.1% | 9.1% | 10.7% | 27.2% | |
| Acidosis | 11.2% | 8.4% | 9.4% | 18.1% | 31.5% |
| Hyperphosphatemia | 7.2% | 7.4% | 9.2% | 9.3% | 23.0% |
| Hyperalbuminemia | 1.0% | 1.3% | 2.8% | 9.0% | 7.5% |
| Hyperparathyroidism | 5.5% | 9.4% | 23.0% | 44.0% | 72.5% |
Note that modification of prevalence according to albuminuria categories data is not yet available to inform this table adequately, though there are limited data to suggest increasing prevalence of hypoalbuminemia, hypertension, anemia, and acidosis as albuminuria category increases.
CKD: chronic kidney disease; GFR: glomerular filtration rate.
Adapted from KDIGO 2012.22
HF is a clinical syndrome defined on the basis of a combination of typical symptoms and signs supported by objective evidence of a structural or functional abnormality of the heart, which is unable to exert its pump function in an effective manner (Table 5).33 HF results in failure to deliver oxygen at a rate commensurate with the requirements of the metabolizing tissues, despite normal filling pressures (or only at the expense of increased filling pressures).15,17,33Table 6 presents the typical symptoms and signs of HF.33
Diagnostic criteria of heart failure with reduced or preserved ejection fraction.
| The diagnosis of HF with reduced EF requires three conditions to be satisfied: |
| 1. Symptoms typical of HF |
| 2. Signs typical of HF |
| 3. Reduced LVEF |
| The diagnosis of HF with preserved EF requires four conditions to be satisfied: |
| 1. Symptoms typical of HF |
| 2. Signs typical of HF |
| 3. Normal or only mildly reduced LVEF and LV not dilated |
| 4. Relevant structural heart disease (LV hypertrophy/LA enlargement) and/or diastolic dysfunction |
EF: ejection fraction; HF: heart failure; LA: left atrial; LV: left ventricular; LVEF: left ventricular ejection fraction.
Adapted from McMurray et al.33
Typical symptoms and more specific signs of heart failure.
| Symptoms | Signs |
|---|---|
| Breathlessness Orthopnea Paroxysmal nocturnal dyspnea Reduced exercise tolerance Fatigue, tiredness, increased time to recover after exercise Ankle swelling | Elevated jugular venous pressure Hepatojugular reflux Third heart sound Laterally displaced apical impulse Cardiac murmur |
Adapted from McMurray et al.33
HF is estimated to affect 2-3% of people worldwide, rising to ≥10% among those 70 years or older,14 and is the leading cause of hospitalization in patients over the age of 65. It is a global health issue that represents a substantial financial burden on healthcare systems and leads to high hospitalization rates and mortality (around 50% of people diagnosed with HF will succumb within five years of diagnosis).14,34–37
Cardiovascular disease and CKD share many risk factors. The major cardiovascular risk factors are diabetes, hypertension, dyslipidemia, obesity and smoking; all of these are also associated with the development of CKD.32 Although cardiovascular morbidity and mortality have markedly decreased in recent years in the general population, their rates remain high among CKD patients.38
Definition, diagnosis, biomarkers, prognosis and treatment of type 4 cardiorenal syndromeThe heart is highly dependent on the regulation of sodium and water homeostasis by the kidneys, which in turn directly depend on blood flow and pressure generated by the heart.39 Accordingly, not only does cardiac compromise affect the renal system, but primary renal dysfunction can trigger modifications that affect the cardiovascular system.39,40 Functional deterioration of either organ, accompanied by structural changes, initiates a cascade of events that leads to and amplifies failure in both organs.13,39 These bidirectional interactions between the heart and kidneys in the context of simultaneous impairment of each organ are what is referred to as CRS.2
Type 4 CRS, also known as chronic renocardiac syndrome, is a subtype in which primary CKD, such as diabetic nephropathy, chronic glomerular disease or autosomal dominant polycystic kidney disease, promotes the progression of chronic HF (with preserved or reduced ejection fraction [EF]), ventricular hypertrophy, diastolic dysfunction, and increased risk of adverse cardiovascular events.8,38,41
CRS is a cycle of organ dysfunction.38 Type 4 CRS is associated with particularly high morbidity and mortality and its incidence is increasing, in part due to increased life expectancy and an aging population. There will thus be more people with cardiac and/or renal dysfunction and these patients will live longer, leading to progression of organ dysfunction and the development of CRS.25,28,42 CKD is a well-known independent cardiovascular risk factor and cardiovascular disease accounts for 50% of deaths in CKD patients.18,24,38 CKD patients are more likely to die prematurely from accelerated cardiovascular disease than from the consequences of renal failure itself: only stage 5 CKD patients have ESRD as the most likely outcome.18,24,32
There is no single test able to diagnose type 4 CRS. The most common diagnostic criteria are the combination of underlying CKD with concomitant cardiovascular pathology.43 It should be noted that the mere coexistence of cardiac and renal disease is not sufficient for a diagnosis of type 4 CRS: CKD must causally underlie the occurrence of chronic HF, supported by both temporal association and pathophysiological plausibility.17 Moreover, the available information on type 4 CRS comes mainly from retrospective studies, in which it is difficult to clearly discriminate the primary from the secondary pathological process and so to determine the epidemiology of type 4 CRS. The ADQI consensus conference recommended that these patients be classified as having both types 2 and 4 CRS.3,44
When cardiac and renal dysfunction occur, it is important to identify biomarkers to detect early functional and structural damage and to risk stratify patients by examining cardiac and renal parameters to enable individualized treatment and prognosis.4,7,25 These biomarkers may facilitate early diagnosis, prognosis, targeted intervention and monitoring of CRS progression (Tables 7 and 8).6,7,45 They can be any measurable parameter, like components of serum or urine or variables from imaging studies.6 In patients with CRS, a group of multiple biomarkers, rather than a single test, may improve diagnosis and better define prognosis.46 However, biomarkers should not replace clinical assessment of CKD patients and should be considered a complement to clinical reasoning.45
Cardiac and renal biomarkers of cardiorenal syndrome.
| Cardiac biomarkers of cardiovascular risk in CKD patients | Renal biomarkers of cardiovascular outcomes in CKD patients |
|---|---|
| Cardiac troponins | Cystatin C |
| Natriuretic peptides: ANP, BNP, NT-proBNP and CNP | Neutrophil gelatinase-associated lipocalin |
| Asymmetric dimethylarginine | N-acetyl-beta-D glucosaminidase |
| Homocysteine | Kidney injury molecule-1 |
| C-reactive protein | Matrix metalloproteinase-9 |
| Serum amyloid-A protein | Interleukin-18 |
| Ischemia-modified albumin | |
| Galectin-3 |
ANP: A-type natriuretic peptide; BNP: B-type natriuretic peptide; CKD: chronic kidney disease; CNP: C-type natriuretic peptide; NT-proBNP: N-terminal pro-brain type natriuretic peptide.
Characteristics of the most promising biomarkers of type 4 cardiorenal syndrome.
| Biomarkers | |
|---|---|
| Cardiac troponins | • Elevated levels are associated with increased mortality in all stages of CKD.6 • Elevated levels have been observed in CKD patients in the absence of myocardial ischemia: reduced renal clearance, myocardial remodeling, uremic pericarditis and myocarditis may contribute to this elevation.6,7 • Levels have prognostic value (inverse correlation).47 |
| Natriuretic peptides | • ANP inhibits cardiac hypertrophy caused by angiotensin II or endothelin-1 and its anti-fibrotic effects have been reported in cardiac fibroblasts, inhibiting cell proliferation and collagen synthesis induced by TGF-β.48 • BNP has been confirmed as an independent predictor of cardiovascular morbidity and mortality in the general population and in CKD patients.25 • BNP has been shown to be valuable in stratifying cardiovascular risk in CKD patients.48 • BNP reflects myocardial wall stress and has prognostic value in dialysis patients.32,48 • CNP attenuates cardiac hypertrophy and inhibits myocardial interstitial fibrosis induced by angiotensin II.48 • CNP inhibits inflammatory responses within the vascular wall and promotes angiogenesis.48 • CNP has demonstrated anti-proliferative and anti-fibrotic properties and has emerged as a new biomarker of renal structural and functional impairment.49 |
| NT-proBNP | • A marker of LV dysfunction and HF.24 Correlates with neurohormonal deterioration.50 |
| Galectin-3 | • Can bind directly to cardiac fibroblasts and reduce LV function through an increase in collagen production.30 • An independent predictor of cardiovascular mortality; direct correlation between serum levels and markers of chronic inflammation.30 |
| Cystatin C | • Directly involved in the atherosclerotic process and associated with increased LV mass and concentricity.24 |
| Neutrophil gelatinase-associated lipocalin | • A marker of tubular damage that independently predicts CKD progression.50,51 • Correlated with disease severity, independently of the coexistence of renal injury, and with NT-proBNP levels.24,52 |
| Kidney injury molecule-1 | • A marker of tubular damage.46,51 |
ANP: A-type natriuretic peptide; BNP: B-type natriuretic peptide; CKD: chronic kidney disease; CNP: C-type natriuretic peptide; CRS: cardiorenal syndrome; HF: heart failure; LV: left ventricular; NT-proBNP: N-terminal pro-brain type natriuretic peptide; TGF-β: tumor growth factor-beta.
The management of CRS is challenging because of the complex crosstalk between heart and kidneys and because many of the drugs used to control the failure one organ can worsen the other organ's function.10,37 Hence, the treatment of type 4 CRS requires a comprehensive and combined approach, considering the potential pathological mechanisms.38,43 Currently, the treatment of type 4 CRS is mainly based on correction of traditional and non-traditional cardiovascular risk factors, and prevention of CKD progression.5,26 Once expressed, heart disease should be managed according to current guidelines on the treatment of HF, such as those of the European Society of Cardiology.
Pathophysiology of type 4 cardiorenal syndromeTraditional cardiovascular risk factors are highly prevalent in CKD patients. However, these are insufficient to explain the high incidence of cardiovascular complications in the CKD population, which suggests there are still missing links.27,37 The contribution of CKD-specific risk factors should be taken into account, since they are associated with both cardiac-specific events and CKD-related syndromes (Table 9).19,26,33,34,37,53 This review will focus exclusively on non-traditional cardiovascular risk factors, which are being increasingly explored.40
Cardiac-specific events and chronic kidney disease-related syndromes in type 4 cardiorenal syndrome.
| Cardiac-specific events | CKD-related syndromes | |
|---|---|---|
| Heart failure Myocardial infarction Ventricular arrhythmia Hospitalization Cardiac surgery Mechanical support device Death | MIA syndrome | • Malnutrition and chronic inflammation, with activation of protein catabolism and increased synthesis of inflammatory cytokines, contributing to atherosclerotic damage and increased cardiovascular mortality. • Persistent inflammation contributes to atherosclerosis, with endothelial dysfunction and vascular calcification, ultimately leading to cardiovascular complications. |
| CRAS | • Individuals with anemia and hyporesponsiveness or resistance to erythropoietin have a particularly high risk of cardiovascular disease and show reduced survival. • Overtreatment with erythropoiesis-stimulating agents aimed at Hb ≥13 g/dl can also increase the risk of cardiovascular disease. | |
| CKD-MBD | • Mineral and bone disorders include hyperphosphatemia, bone abnormalities and vascular calcification, which in turn contribute to atherosclerosis and cardiac disease. | |
Both traditional and non-traditional cardiovascular risk factors and other biological mechanisms play various roles in the stages of CKD at different times, ultimately resulting in morphological, functional and electrical changes in the myocardium and coronary arteries. Type 4 CRS patients typically present earlier onset and more severe cardiovascular disease, that is more difficult to treat, and worse prognosis. Table 10 summarizes data on cardiac disease in CKD patients (arrhythmias, HF, CAD and sudden cardiac death [SCD]) that define the pathophysiological background characterizing the interaction between the diseased kidneys and the heart, as well as potential targets and clinical manifestations.26,54–57
Clinical and biological data on cardiac diseases in patients with chronic kidney disease.
| Congestive HF and myocardial fibrosis | CAD | Arrhythmias and SCD |
|---|---|---|
| • ECG abnormalities (reduced EF, increased end-systolic and end-diastolic LV diameters and volumes) are frequently reported in all CKD stages.26 • ESRD patients develop cardiac fibrosis, mainly endocardial and epicardial.26 • Most dialysis patients have pathological findings such as LV hypertrophy and LV dilation.26,54 • Congestive HF is associated with systolic failure, LV hypertrophy and CAD.55 • In ESRD patients, congestive HF is an independent adverse prognostic indicator.55 • CKD patients are frequently hypertensive and more likely to have cardiomyopathies and vascular calcification in addition to atherosclerosis.55 | • CKD patients have increased rates of atherosclerotic CAD, higher prevalence of multivessel disease and ECG evidence of previous ischemia.26 • CRP, IL-6 and TNF-α are higher in ESRD patients than in the normal population.26 • Vascular calcification in the media of the vascular wall (Monckeberg's sclerosis) is typical of CKD patients, in contrast to intimal calcification in diabetic and CAD patients.26 • Patients with CACS >10 have lower CFR and worse cardiovascular outcomes.26 • ESRD patients demonstrate a close relationship between MIA syndrome and epicardial adipose tissue density and atherosclerosis.26 • In ESRD patients, dobutamine stress echocardiography is the gold standard for CAD screening in renal transplantation candidates.56 | • Dialysis patients are more prone to develop arrhythmias, especially atrial fibrillation and ventricular tachyarrhythmias.26 • Electrolyte abnormalities, CAD, hypertension, HF and LV hypertrophy are all predisposing factors for arrhythmias.26 • Atrial fibrillation is closely related to CKD stage (odds ratio for atrial fibrillation: 2.20 in stage 1-2 CKD; 1.51 in stage 3 CKD; 2.86 in stage 4-5 CKD).26 • Extreme shifts of electrolytes and changes in blood pressure and/or volume are common in intra- and inter-dialytic periods, leading to mechanical and electrical dysfunction of myocardial cells.26,57 • Almost 50% of cardiovascular deaths in ESRD patients are related to cardiac arrhythmias or SCD.26,57 • Cardiac autonomic dysfunction is related to LV hypertrophy in ESRD patients.57 |
CACS: coronary artery calcification score; CAD: coronary artery disease; CFR: coronary flow reserve; CRP: C-reactive protein; ECG: electrocardiographic; EF: ejection fraction; ESRD: end-stage renal disease; GFR: glomerular filtration rate; HF: heart failure; IL: interleukin; LV: left ventricular; MIA: malnutrition-inflammation-atherosclerosis; SCD: sudden cardiac death; TNF-α: tumor necrosis factor-alpha.
Although type 4 CRS has been intensively studied, its pathogenesis remains incompletely understood.41 The interactions between the heart and kidneys are complex and probably multifactorial in nature.1Figure 2 illustrates the proposed biological pathways involved in type 4 CRS at each CKD stage and the pathophysiological consequences.24,26,53Figure 3 illustrates CKD-related syndromes and their interaction.53
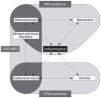
 CKD)-related events occurring at each
CKD)-related events occurring at each  CKD-MBD interact with each other, with inflammation playing a central role in all three mechanisms.
CKD-MBD interact with each other, with inflammation playing a central role in all three mechanisms. Chronic kidney disease-related syndromes. MIA syndrome, CRA syndrome and CKD-MBD interact with each other, with inflammation playing a central role in all three mechanisms. CKD-MBD: chronic kidney disease-related mineral and bone disorders; CRA: cardiorenal anemia; MIA: malnutrition-inflammation-atherosclerosis.
Albuminuria is one of the strongest non-traditional cardiovascular risk factors. Epidemiological studies have established that high urinary albumin excretion is predictive of cardiovascular morbidity and mortality independently of GFR, which suggests that albuminuria and reduced GFR have distinct mechanisms as cardiovascular risk factors.20,37 Cardiovascular risk increases progressively with deterioration of renal function and with the degree of albuminuria; even microalbuminuria is significantly associated with cardiovascular disease.20,37 The mechanisms underlying this association are still unknown.20
Microalbuminuria results from glomerular injury or reduced tubular reabsorption of filtered albumin. Macroalbuminuria is an indicator of intrinsic kidney disease, a predictor of CKD progression and a mediator of renal injury.20,37
Iron disorders and anemiaAnemia is common in CKD patients, but it is unclear whether it is only a marker or also a mediator of CRS progression.38,40,41 In type 4 CRS, anemia is primarily due to deficient erythropoietin production; erythropoietin also prevents apoptosis and increases the number of cardiac and renal cells.38,41,58 Iron deficiency has also been shown to be an independent predictor of unfavorable clinical outcomes. This finding indicates that the interaction between iron deficiency and anemia is critical for the pathogenesis and prognosis of type 4 CRS, through the action of hepcidin.41 Since hemoglobin is an antioxidant, anemia may also contribute to an abnormal oxidative state. Moreover, anemia is one of the most important factors for the development of left ventricular (LV) hypertrophy, exacerbating the cardiac effects of hypertension in CKD patients.15,38
Mineral and bone disordersMineral and bone homeostasis, maintained through interactions between calcium, phosphorus, parathyroid hormone (PTH), vitamin D and fibroblast growth factor (FGF)-23 (FGF23), is commonly disrupted in CKD patients (especially with GFR <45 ml/min/1.73 m2), and mineral and bone disorders are frequently associated with type 4 CRS.32,38,43 Mineral homeostasis disorders lead to demineralization of bone and excessive calcification of soft tissue: CKD patients cannot excrete phosphorus, which results in hyperphosphatemia and raised PTH, reduced production of the active form of vitamin D (1,25-dihydroxycholecalciferol), and hypocalcemia.32,43,59,60 Together, hyperphosphatemia and hypocalcemia can precipitate arrhythmias and depress cardiac contractility.6 Hyperphosphatemia is also associated with increased vascular wall stiffness and lower vascular compliance, and is thus strongly associated with cardiovascular disease in CKD patients.24,38,40,43,59 Moreover, these patients present high PTH levels, which are associated with adverse outcomes.38
Vitamin D is involved in many physiological processes, especially mineral and bone homeostasis. 1,25-dihydroxycholecalciferol is synthesized in the kidneys and exerts its biological effects by binding to the vitamin D receptor (VDR).38,41,60 The ubiquitous distribution of the VDR is responsible for its pleiotropic effects when activated (Figure 4).38,41,59–61 1,25-dihydroxycholecalciferol deficiency has been identified as a cardiovascular risk factor. At the same time, CKD patients show lower 1,25-dihydroxycholecalciferol levels than the general population.60 Impaired 1,25-dihydroxycholecalciferol synthesis can lead to vascular calcification and atherosclerotic CAD (Figure 5).30,62 Furthermore, VDR activation functions as a negative regulator of the renin-angiotensin-aldosterone system (RAAS), which when insufficiently activated may contribute to type 4 CRS.38,41,59,60 Nargesi et al. concluded that serum vitamin D is independently associated with future hard CAD events (Figure 6).63
 PTH: parathyroid hormone;
PTH: parathyroid hormone; Metabolism and actions of vitamin D that may be implicated directly and indirectly in the atherogenic process. Mechanisms with demonstrated clinical significance are marked in yellow. 1-OHase: 1α-hydroxylase; 25-OHase: 25-hydroxylase; HDL: high-density lipoprotein; ICAM-1: intercellular adhesion molecule-1; IL: interleukin; LDL: low-density lipoprotein; PTH: parathyroid hormone; RAAS: renin-angiotensin-aldosterone system; TNF-α: tumor necrosis factor alpha; VCAM-1: vascular adhesion molecule-1.
 CAD: coronary artery disease;
CAD: coronary artery disease; Impact of vitamin D deficiency on cardiovascular risk. CAD: coronary artery disease; PTH: parathyroid hormone; RAAS: renin-angiotensin-aldosterone system; SNS: sympathetic nervous system; VDR: vitamin D receptors.
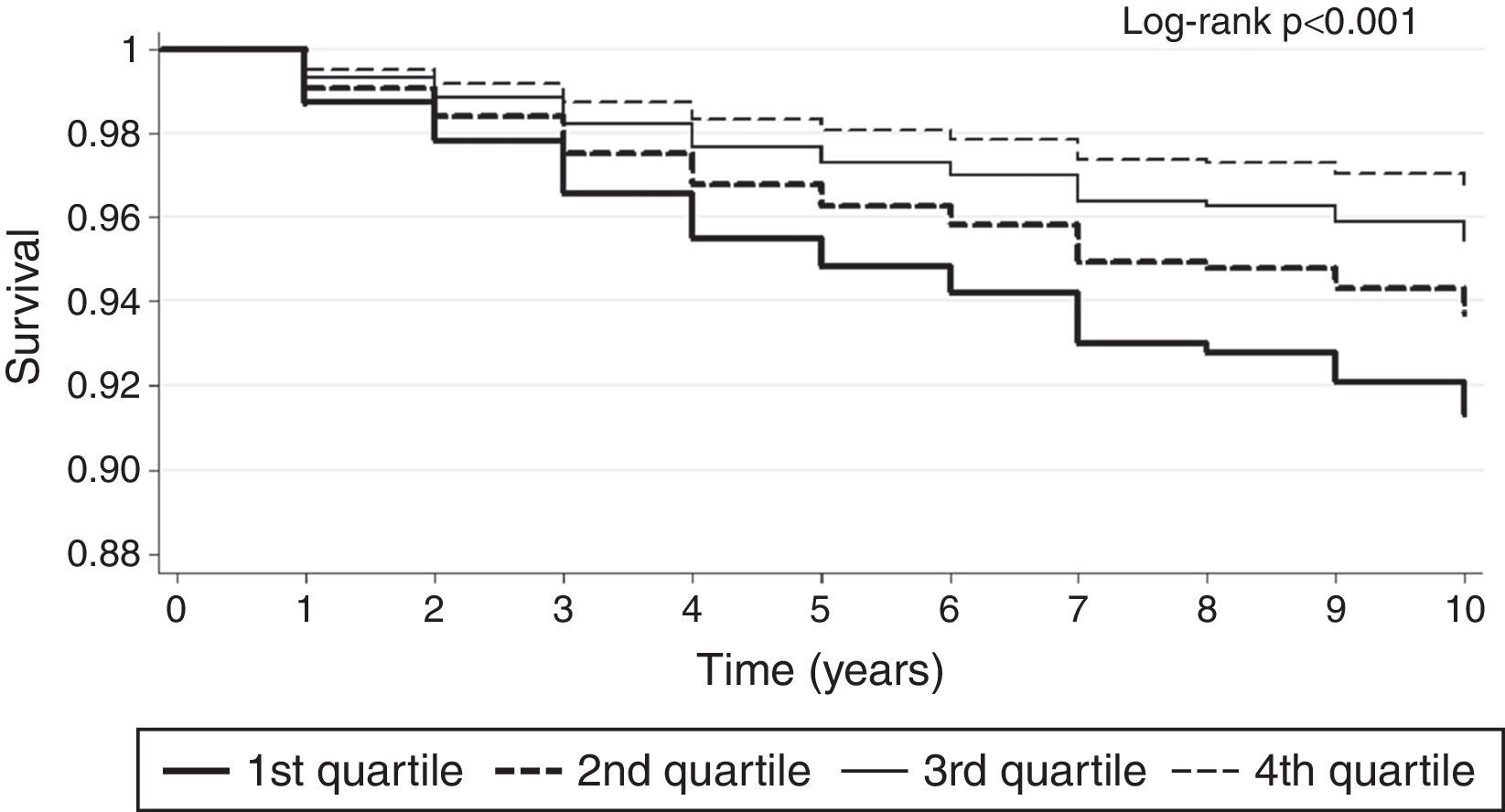
Kaplan-Meier survival plot of patients in quartiles of vitamin D level.
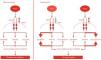
FGF23 is a newly discovered hormone produced in the bone that regulates phosphate and vitamin D metabolism by the kidneys and is a powerful predictor of morbidity and mortality in patients with CKD and ESRD. FGF23's main physiological functions are mediated by FGF receptors, generally in the presence of Klotho coreceptors.59 Decreased phosphorus excretion triggers FGF23 production, which in turn stimulates Klotho coreceptors in the kidneys.8,30 The decline in Klotho expression with CKD progression leads to compensatory elevation of FGF23 levels, resulting in typical CKD manifestations, such as hyperphosphatemia, secondary hyperparathyroidism and bone disease, and progression to ESRD. FGF23 level is directly associated with LV hypertrophy and cardiac remodeling, possibly via a direct effect on cardiomyocytes (Figure 7).27,30,59,64
 FGF2 and
FGF2 and The effects of FGF2 and FGF23. In the kidneys and parathyroid glands, FGF23 signaling requires FGFR and the Klotho coreceptor. FGF23-Klotho binding to FGFR induces signaling through three major pathways. FGF23 regulates phosphorus balance by altering expression of genes involved in mineral and bone metabolism. In cardiomyocytes, FGF2 signaling requires FGFR and HSP as coreceptor and signals primarily through the Ras-MAPK pathway. Binding of FGF23 to FGFR on cardiomyocytes stimulates autophosphorylation of the receptor tyrosine kinase independent of Klotho, which is not expressed in cardiomyocytes, and signals primarily through the PLCγ-calcineurin pathway. FGF2: fibroblast growth factor 2; FGF23: fibroblast growth factor 23; FGFR: fibroblast growth factor receptor; HSP: heparan sulfate proteoglycans; LV: left ventricular; PLCγ: phospholipase C-gamma.
As CKD evolves, there is progression of parenchymal and glomerular sclerosis and, ultimately, uremia. This entity is characterized by the accumulation of uremic toxins and inflammatory cytokines in the blood, resulting in oxidative stress and consequent accelerated atherosclerosis and CKD progression.24,39 Uremic toxins can be divided into three major groups: small water-soluble compounds; protein-bound compounds; and large compounds. Most small-molecule uremic toxins are removable by conventional dialysis; they are generally not problematic.31,65 By contrast, protein-bound uremic toxins (PBUT) are poorly removed by current dialysis techniques because of their size. In particular, indoxyl sulphate (IS) and p-cresyl sulphate are recognized as cardiovascular risk factors, as they stimulate inflammation, fibrosis, endothelial dysfunction, vascular smooth muscle cell proliferation, vascular calcification, accelerated atherosclerosis and consequent cardiovascular disease.24,30,37,65 Accordingly, it has been suggested that PBUT play an important role in the development of CRS, explaining the relatively poor prognosis of these patients.65 IS appears to be the strongest evidence-based PBUT involved in the development of types 2 and 4 CRS.31 Homocysteine is a PBUT that adversely affects the heart and kidneys in the setting of type 4 CRS. The negative effect of the hostile uremic milieu is highlighted in findings that demonstrated favorable outcomes on left ventricular (LV) abnormalities and cardiac death in a frequent dialysis group, as compared to a conventional dialysis group.43 It should be noted that most studies on CRS merely document an association and no reports of direct evidence of renal or cardiac dysfunction induced by PBUT have yet been published.39
The role of the uremic milieu in the development of CRS still needs to be appraised; specific PBUT or combinations thereof could directly cause metabolic and physiological derangements and contribute to progression of type 4 CRS.39
Left ventricular abnormalitiesLV abnormalities are strongly associated with adverse outcomes in ESRD patients. Echocardiographic studies show three patterns of cardiomyopathy affecting up to 85% of ESRD patients: LV hypertrophy (50-80%); LV dilatation (20-40%); and LV systolic dysfunction (16%). LV hypertrophy develops early in CKD, as an adaptive phenomenon in response to increasing pressure and volume. However, over time, it becomes maladaptive, leading to cardiac dysfunction.25 LV hypertrophy is considered an independent predictor of cardiovascular mortality.25,27 Uremic cardiomyopathy also contributes to further activation of neurohormonal pathways with detrimental effects.43 Dialysis itself may not directly contribute to LV hypertrophy or be able to reverse long-standing hypertrophic changes, but its inability to clear specific uremic toxins could indirectly contribute to cardiac remodeling.30 In patients with CKD, the major determinant of LV hypertrophy is hypertension, but anemia, abnormal mineral and bone metabolism and arteriosclerosis all contribute to LV hypertrophy, accompanied by increased wall thickness and reduced ventricular compliance.25,30
Cardiac remodeling is seen even in the early stages of CKD.32 ESRD in type 4 CRS is characterized by recruitment and activation of T cells and cardiac macrophages, which direct the cycle of cardiomyocyte apoptosis and replacement fibrosis, presenting an abnormal myocardial architecture. Histopathological changes may include LV hypertrophy, decreased capillary density and myocardial perfusion, myocardial fibrosis, and calcification of soft tissue, including vascular structures and heart valves.8,32 These changes constitute what has been termed ‘CKD cardiomyopathy’, and have been associated with systolic and diastolic dysfunction, chronic edema and increased rates of HF hospitalization and death.8
End-stage renal diseaseWith CKD progression, the renal parenchyma, already presenting inflammation and glomerular and interstitial damage, begins to display fibrotic and sclerotic symptoms. At the same time, the clinical picture evolves to overt uremia and ESRD, and renal replacement therapy becomes mandatory.38
Hemodialysis (HD) and peritoneal dialysis (PD) themselves contribute to cardiovascular morbidity and mortality, due to inflammation and hemodynamic stress induced in these patients.24,66 Different factors may lead to a chronic inflammatory state, including blood-membrane interaction, blood-catheter interaction, catheter infection, and dialysate contaminants (Figure 2).24,26,53 Most HD patients die from SCD, attributable mostly to sudden arrhythmic events and HF.15,66 PD also increases cardiovascular risk in CKD patients, but further long-term studies assessing cardiovascular outcomes in PD patients are required.24,38
Biological and hemodynamic mechanismsThe mechanisms that underpin CRS are probably multifactorial and may include neurohormonal activation and hemodynamic alterations, inflammation, oxidative stress, endothelial dysfunction, and endoplasmic reticulum (ER) stress (Figure 2).16,24,26,41,53 Interactions have been reported between these pathways, which are common to both heart and kidney failure.13,16 Their permanent activation plays a central role in the initiation and progression of CRS, at least in part.14,16 In CRS patients, a plethora of risk factors also contributes to the progression of cardiac and renal dysfunction, and ESRD patients face additional risks related to dialysis procedures.39 These factors in the pathophysiology of type 4 CRS will be covered in more detail in the following sections.
Neurohormonal activation and hemodynamic alterationsNeurohormonal activation and related hemodynamic alterations play a critical role in all types of CRS.37 Over time, neurohormonal systems (including the RAAS, the sympathetic nervous system [SNS], and the arginine vasopressin and endothelin systems) have evolved to ensure homeostasis. These systems, which increase fluid retention, re-establish cardiac output and reduce renal blood flow, are essential for survival in heart failure.20,37,67 Although initially appropriate and compensatory, sustained activation of these mechanisms ultimately becomes detrimental and results in a progressive decrease in cardiac and renal performance.28,48,68–70 Neurohormones are also strong triggers of oxidative injury and act in concert with other factors to accelerate organ damage.28,37 At the same time, systems that promote fluid excretion and increase renal blood flow, such as natriuretic peptides, are blunted even if their levels are increased, which aggravates fluid retention and cellular injury.67 Loss of renal mass resulting from CKD leads to the accumulation of sodium and water. In patients with CRS, the resulting hypertension and fluid overload, coupled with the direct effects of angiotensin II and aldosterone on cardiomyocytes, are responsible for a hypervolemic state, increasing cardiac work, with compensatory LV hypertrophy and cardiac fibrosis.24,30,39
The RAAS and SNS are both key regulatory systems for the maintenance of cardiovascular and renal function, interacting to induce systemic vasoconstriction and sodium retention.32,41 They are common to both renal and cardiovascular failure and may amplify each other, which explains their synergistic effects that magnify the poor outcomes of either disease alone.1,16,41,68
The role of the RAAS in CRS is complex.70 Angiotensin II is the primary bioactive peptide in the RAAS and acts via the angiotensin type 1 receptor to exert its intracellular effects.71 When excessively activated, the RAAS is associated with increased inflammation, fibrosis, oxidative stress (through activation of NADPH oxidase, with increased formation of reactive oxygen species [ROS]) and endothelial dysfunction, thereby resulting in progression of CRS.28,37,41,68 This prolonged activation also leads to sodium retention with additional volume overload and cardiac remodeling, thus perpetuating the vicious circle.14,37,43
As CKD progresses and GFR decreases, the SNS is overstimulated as a result of renal ischemia, suppression of nitric oxide (NO) synthesis and maladaptive activation of the RAAS, among other causes. Excessive SNS activation can cause reduced myocardial beta-adrenergic receptor density, particularly beta-1, as well as uncoupling of the receptor from intracellular signaling mechanisms; cardiac remodeling; LV abnormalities, culminating in cardiomyocyte dysfunction and fibrosis (‘CKD cardiomyopathy’); vasoconstriction; insulin resistance and dyslipidemia, exacerbating atherosclerosis; inflammation; and activation of the RAAS, by directly stimulating renin release.40,43,66,68 Note that, in CKD patients, these deleterious effects are exacerbated due to reduced catecholamine clearance, since renal function is already impaired.69
InflammationThere is increasing evidence regarding the role of the inflammatory response in CRS.41 In both cardiac and renal dysfunction, there is a state of permanent inflammation, which affects endothelial function and lipid profile.32 Patients with chronic HF and CKD produce various inflammatory cytokines, including tumor necrosis factor-alpha (TNF-α), transforming growth factor-beta (TGF-β), interleukin (IL)-1, and IL-6, which regulate cell survival and death.2,37,40 TGF-β is thought to be the primary driver of kidney fibrosis and parenchymal loss.18 In CRS patients, the production of these inflammatory mediators is much higher than in the general population and is associated with LV remodeling, cardiomyocyte apoptosis, LV dysfunction, pulmonary edema, anorexia, cachexia and renal failure.40,41 However, the biological processes driving this chronic inflammatory state are not entirely understood.71 The origin of inflammation in CKD is multifactorial.40 There is evidence that activation of the RAAS and SNS, due to decreased renal filtration capacity, promotes an inflammatory response.41,71 Several studies suggest that volume overload and venous congestion (and venous pressure) play an important and deleterious role in CRS patients, as an additional source of inflammatory mediators; in fact, evidence suggests that venous congestion, not arterial blood flow, is one of the most significant mediators of CRS.14,41,68,69 Oxidative stress has also a central role in type 4 CRS, exerting its detrimental effects through inflammatory mediators and simultaneously being enhanced by the typical chronic inflammatory state of these patients.14,24,41 Uremia and other comorbidities, such as diabetes, may also contribute to high levels of inflammatory cytokines.25 At the same time, reduced clearance of these cytokines can itself also contribute to the characteristic low-grade inflammatory state of type 4 CRS.25 Systemic inflammation has harmful biological effects on the cardiac and renal systems, predisposing them to functional and structural damage (Figure 8).71,72 It appears to be a crucial part of CRS progression.36,71
 ROS are strongly implicated in the pathological signaling leading to
ROS are strongly implicated in the pathological signaling leading to Development of chronic kidney disease and chronic inflammation: a vicious circle. ROS are strongly implicated in the pathological signaling leading to CKD, such as RAAS activation. Angiotensin II can activate the NF-kB pathway, which is involved in the control of the transcription of a variety of cellular genes that regulate the inflammatory response by the production of cytokines. In addition, angiotensin II may cause renal fibrosis by production of TGF-β and the induction of extracellular matrix proteins such as type I procollagen, fibronectin and type IV collagen. CKD: chronic kidney disease; iNOS: inducible nitric oxide synthase; RAAS: renin-angiotensin-aldosterone system; ROS: reactive oxygen species; TGF-β: tumor growth factor-beta.
In patients with CRS, there is an imbalance between ROS and NO toward oxidative stress, due to increased ROS production and decreased NO synthesis.31,37 ROS are produced as a result of redox reactions in various cells and have been recognized as key chemical mediators causing cellular aging, increased production of inflammatory cytokines such as TNF-α, TGF-β, IL-1 and IL-6, and organ dysfunction in both cardiac and renal disease.14,37,40 Increased ROS production may be induced by inappropriate activation of the RAAS and, consequently, decreased NO availability (due to inactivation of NO by ROS), which impairs vasodilation, reduces renal perfusion and allows platelet aggregation and neutrophil adhesion.31,40 Increases in ROS are also linked to uremic cardiomyopathy. Endothelial damage caused by oxidative stress, in turn, enhances intimal accumulation of oxidized low-density lipoprotein, the initiating event of atherosclerosis.31 Oxidative stress may thus represent a final common pathway for cardiac and renal failure.40,73 In comparison with controls controlled for age, gender and weight, oxidative stress levels were significantly higher in type 4 CRS patients and significantly correlated with cardiac remodeling and reduced cardiac function.28 Interactions between inflammation, fibrosis, endothelial damage, neurohormonal activation, uremia and atherosclerosis may thus potentiate CRS through excessive oxidative stress.31,40 Several factors thought to enhance oxidative stress (such as angiotensin II, endothelin-1 and TGF-β1) can upregulate fibroblast growth factor-2 (FGF2) and be suppressed by antioxidants. Given that FGF2 promotes myocardial hypertrophy and fibrosis, the harmful effects of oxidative stress on the heart in type 4 CRS may occur partially through this mediator.28
Oxidative stress can be monitored with oxidative stress indicators such as malondialdehyde, superoxide dismutase and asymmetric dimethylarginine, and advanced oxidation protein products.28
Endothelial dysfunctionThe endothelium plays a central role in the atherosclerotic process and in the regulation of cardiac and renal function, and so endothelial damage can contribute to dysfunction of both organs, to cardiovascular mortality (even in the early stages of CKD) and to progression of renal disease.14,32 Reduced NO bioavailability is a major pathophysiological component of endothelial dysfunction. NO synthesis by the endothelium is critically dependent on the transport of its substrate, L-arginine, via the cationic amino acid transporter-1. Impairment of this pathway has been demonstrated individually in both cardiac and renal failure, leading to accelerated atherosclerosis, albuminuria and decreased vasodilation response.14,37
Endoplasmic reticulum stressSynthesis of transmembrane and secretory proteins occurs within the ER and is extremely important for the normal functioning of both the heart and kidney. Dysregulation of protein synthesis and processing within the ER causes accumulation of unfolded proteins, thereby leading to ER stress and activation of the unfolded protein response. Severe CKD may bring about hemodynamic changes through plasma volume expansion that result in cardiomyocyte hypertrophy. This process appears to necessitate an increase in protein synthesis, which directly results in ER stress. Although ventricular hypertrophy may be initially adaptive, prolonged or severe ER stress can lead to cardiomyocyte apoptosis. ER stress may also result from the myocardial reaction to hormones such as renin, angiotensin II and arginine vasopressin. Renal failure attributable to proteinuria and uremia also induces ER stress within the kidney, which leads to the transformation of tubular epithelial cells, fibrosis and tubular cell apoptosis, worsening renal function.74 Consequently, CRS may develop into a vicious circle of cardiac and renal impairment with an unfavorable prognosis.68,74
Mitochondria are the major sources of ROS. Oxidative reactions are essential for cellular function; however, oxidative stress is also an important factor in accelerating heart or kidney dysfunction.41 Decreased mitochondrial oxidative metabolism has been reported in cardiomyocytes during HF. A thorough understanding of the cellular and mitochondrial pathways involved in both cardiac and renal oxidative stress is essential in order to enable the development of novel and more effective therapies able to improve survival and prognosis in CRS patients.11,68
ConclusionsThe ADQI consensus conference proposed a definition and classification of CRS, aiming for a better delineation of each subtype. Although the exact pathophysiology of type 4 CRS is not completely understood, the mechanisms involved are probably multifactorial and may include neurohormonal activation and hemodynamic alterations, inflammation, oxidative stress, and endothelial dysfunction.16,40 CKD patients are particularly predisposed to cardiac dysfunction, due to the high prevalence of traditional cardiovascular risk factors in this population, but the contribution of CKD-specific risk factors should also be taken into account.15,19,27,37 Both traditional and non-traditional risk factors accelerate atherosclerosis in CKD patients.20,24,42 There is growing evidence that oxidative stress is a major connector in the development and progression of type 4 CRS, contributing to CKD progression, increased inflammation, endothelial dysfunction, accelerated atherosclerosis and cardiac remodeling.13,24,28,40,42,66,73 This series of events characterizes type 4 CRS, in which patients are at higher risk for LV hypertrophy, HF with preserved or reduced EF, hypertensive cardiomyopathy, CAD, arrhythmias and SCD.24,26,54,55 Giving its complexity, worse prognosis and increasing incidence, type 4 CRS is becoming a significant public health problem.6,24,37,47 There is no single test able to diagnose CRS.43 Several new biomarkers are being studied aiming for the development of useful tools for early diagnosis and accurate prognosis in these patients.4,50 Although there has been significant progress in the discovery of biomarkers, further trials are warranted to establish their exact role and to validate them in patients with type 4 CRS, before their translation into clinical practice.7,32
Much remains to be elucidated about type 4 CRS: despite significant progress over the last decade, there are still significant questions regarding its pathophysiology and there is as yet no specific therapy.41 In-depth understanding of the interactions between the heart and kidneys may provide potential targets for intervention.24 These knowledge gaps in the literature constitute opportunities for future research.44 Finally, we emphasize the need to produce guidelines for targeted prevention and effective therapeutic strategies in type 4 CRS: a better understanding of the mechanisms involved in type 4 CRS may provide potential targets for intervention.24
Conflicts of interestThe authors have no conflicts of interest to declare.
