

Cardiomyopathies may present as a manifestation of various inherited syndromes. Recognizing the rarity and diagnostic challenges of syndromic and metabolic cardiomyopathies is crucial, as their identification holds significant implications for targeted treatment and enables the use of specific risk stratification tools. Genetics has assumed a pivotal role in clarifying the pathophysiology of cardiomyopathies, facilitating molecular diagnosis, and enabling effective family screening. The advent of next-generation sequencing has revolutionized genetic testing, enabling cost-effective, high-throughput analyses, facilitating the diagnosis of these rare conditions, and allowing the provision of specific management and therapeutics.
As miocardiopatias podem-se apresentar como manifestação de diversas síndromes hereditárias. Reconhecer as miocardiopatias sindrómicas e metabólicas como doenças raras e de diagnóstico desafiante é essencial, uma vez que a sua identificação tem implicações significativas para um tratamento individualizado e na utilização de ferramentas específicas para estratificação de risco. A genética assumiu um papel fundamental na clarificação da fisiopatologia das miocardiopatias, facilitando a identificação do diagnóstico molecular e possibilitando o rastreio familiar eficaz. O desenvolvimento da sequenciação de nova geração revolucionou os testes genéticos, permitindo análises de alto rendimento a custos acessíveis, facilitando o diagnóstico destas doenças raras, e permitindo acompanhamento e terapêuticas específicas.
Cardiomyopathies constitute a heterogeneous group of diseases that are characterized by structural and functional abnormalities in the heart muscle and are unexplained by coronary or valvular disease or abnormal loading conditions.1 The category of syndromic and metabolic cardiomyopathies includes several rare conditions such as lysosomal storage diseases, mitochondrial cardiomyopathy, RASopathies, Anderson-Fabry disease (FD), and neuromuscular diseases. These conditions are challenging to diagnose due to their rarity. However, early identification is crucial as it enables targeted treatment and the use of specific risk stratification tools to prevent sudden cardiac events.2,3
Cardiomyopathies are a significant manifestation of certain inherited metabolic disorders, especially lysosomal storage disorders. However, cardiovascular involvement may extend beyond metabolic cardiomyopathy, which can be either dilated or hypertrophic, and may also include arrhythmias and vascular and/or valvular disease. Moreover, the cardiac phenotype may not be the initial symptom; rather, patients may present with extracardiac and multisystem signs and symptoms that can provide essential clues to aid in the clinical diagnosis. Enzymatic and molecular genetic analysis can be used to confirm the diagnosis.3
In this review, we aim to offer clinicians a targeted, evidence-based guide on performing genetic testing as a valuable tool to enhance the management of inherited cardiomyopathies. The review is divided into two parts: the approach to hypertrophic cardiomyopathy (HCM), non-dilated left ventricular (LV) cardiomyopathy, and dilated cardiomyopathy (DCM) (Part 1), and the management of syndromic and metabolic cardiomyopathies and phenocopies (Part 2). This manuscript constitutes Part 2.
Metabolic cardiomyopathiesLysosomal storage disordersThe lysosomal storage disorders are a group of diseases resulting from a deficiency of a single enzyme required for the metabolism of macromolecules, which leads to an accumulation of its substrates inside lysosomes. These diseases are classified according to the type of accumulated molecule, for example glycogen storage diseases (Pompe and Danon) and mucopolysaccharidoses and glycosphingolipidoses (FD).4
Definitive diagnosis is crucial for lysosomal storage disease due to the availability of enzyme replacement therapy, which substantially improves many of the features of the disease, including some aspects of cardiac involvement.5
Among the lysosomal storage disorders affecting the heart, the most prevalent is FD. This rare X-linked recessive disorder presents with a progressive multisystem clinical course. It is caused by deficient or absent activity of lysosomal alpha-galactosidase A, an enzyme encoded by the GLA gene. The enzyme deficiency leads to progressive lysosomal accumulation of the glycolipid globotriaosylceramide in most organs, leading to cell death, inflammation, oxidative stress, and fibrosis. Cardiomyocyte hypertrophy results from increased concentrations of sphingolipid, vascular smooth muscle cell proliferation, and myocyte hypertrophy. Notably, in men, this glycolipid may account for only 1–2% of cardiac mass, with the remaining increase attributed to myocyte hypertrophy.6
Based on residual enzyme activity levels, Fabry disease can be divided into two distinct forms: the classical form and late-onset presentation. The classical form is characterized by early onset and severe clinical presentations and is linked to GLA variants that result in either absent or significantly reduced enzyme activity. Symptoms include acroparesthesias, angiokeratomas, neuropathic pain, hypohidrosis, heightened sensitivity to temperature changes, and gastrointestinal issues. As patients progress into adulthood, the spectrum of manifestations expands to include cardiomyopathy, brady- and tachydysrhythmias, renal failure, cerebrovascular accidents, and sensorineural deafness.7
By contrast, late-onset presentation is linked to GLA variants that yield a degree of residual enzymatic activity (greater than 1%) and are associated with attenuated phenotypes. These presentations primarily involve the emergence of cardiac, renal, and/or cerebrovascular symptoms during adulthood. Although Fabry disease is widespread across the world, some geographical clusters based on founder variants of this late-onset form have been identified, including the p.F113L variants in the Portuguese region of Guimarães.6 In this group of patients, there is frequent cardiac involvement, which is the predominant determinant of prognosis.
During the investigation of HCM, the discovery of subepicardial late gadolinium enhancement (LGE) in the basal inferolateral wall and bifascicular block are red flags for FD. These indicators are notably robust in suggesting the potential presence of the disease. Moreover, the electrocardiogram (ECG) can reveal additional markers such as a short PR interval, electrocardiographic criteria for hypertrophy, and first-degree atrioventricular (AV) block. Additionally, cardiac magnetic resonance imaging (CMRI) can add information by identifying low native T1 mapping values, which reflect elevated lipid content attributed to sphingolipid accumulation.8 As the disease progresses, extracellular volume increases due to hypertrophy and myocardial fibrosis, which can ultimately lead to T1 pseudonormalization.9
A diagnosis of FD is reached through the demonstration of decreased or absent serum or leukocyte alpha-galactosidase A activity or pathogenic variants in the GLA gene. Enzymatic screening utilizing dried blood spots on filter paper has emerged as a straightforward and accessible method.10 However, for females genetic testing is essential for diagnosis. Although the clinical response is not always optimal, enzyme replacement therapy may ameliorate symptoms and renal function, potentially reducing LV hypertrophy (LVH), cardiac events, stroke, and overall mortality.11
Glycogen storage disordersGlycogen storage diseases are inherited genetic disorders that result from the accumulation of glycogen, often presenting in children. These diseases may be associated with HCM, but can also have a restrictive phenotype. These patients typically show extreme LVH and must be differentiated mainly from those with HCM.12
One example is Danon disease, an X-linked dominant disorder that is caused by a deficiency of lysosome-associated membrane protein-2 (LAMP2), which is encoded by the LAMP2 gene. Histologically there is excess glycogen accumulation in cardiomyocytes and skeletal muscle fibers, leading to the formation of vacuoles. The finding of extreme LVH, with wall thickness measurements reaching as much as 65 mm, along with increased QRS voltages and T wave abnormalities on the ECG, are characteristic of Danon disease. As well as skeletal and cardiac muscle involvement, patients with this condition may exhibit mild to moderate cognitive impairment. This is attributed to the high expression of LAMP2 in the brain, in addition to the heart and skeletal muscles. Moreover, the effects of this multisystem disorder may extend to other organs, encompassing retinopathy, gastrointestinal and hepatic complications, and pulmonary manifestations. While LVH is the most prevalent and typical presentation, the phenotype displays gender-based variations due to its X-linked dominant inheritance. As anticipated, the disease tends to manifest in females at a later stage of life. In this case, the predominant symptoms often center around cardiac manifestations; moreover, females may present DCM and asymmetric LVH.13
5′-AMP-activated protein kinase subunit gamma-2 (PRKAG2) cardiomyopathy is a glycogen storage disorder with autosomal dominant inheritance. This condition typically manifests with progressive cardiac hypertrophy, conduction abnormalities, and arrhythmias, with approximately 33% of individuals affected by atrial fibrillation or atrial flutter.14 The spectrum of electrocardiographic abnormalities most commonly includes ventricular pre-excitation, often referred to as Wolff-Parkinson-White syndrome, and bundle branch block. Maximum ventricular wall thickness varies widely between patients, ranging from normal values to over 40 mm. A restrictive filling pattern with progression to DCM is a leading cause of cardiac transplantation or death. CMRI may show LGE only in advanced stages, along with high T1 values caused by fibrosis.15 While the inheritance pattern differs from Danon disease, certain manifestations overlap. However, it is important to note that disease progression is generally less severe than in Danon disease, and muscle weakness and myalgia are rarely reported in PRKAG2-related disorders. Additionally, retinal disease and intellectual impairment are not observed in these disorders, providing a distinguishing feature between the two conditions (Figure 1).

Pompe disease (also known as glycogen storage disease II) is a multisystem disorder with autosomal recessive inheritance that results from a deficiency in acid maltase, a protein encoded by the GAA gene. Onset is usually in infancy, causing massive cardiac hypertrophy in neonatal and pediatric ages, leading to LV outflow tract obstruction. Diastolic dysfunction is generally present and systolic dysfunction may be seen later. Adult-onset acid maltase deficiency is a rare variant of the disease: as it seldom affects the heart, the cardiac phenotype of adults with this condition is poorly characterized. The morphology of the hearts of patients diagnosed with glycogen storage disease type III, also known as Cori disease (caused by pathogenic variants in the AGL gene), usually mimics HCM but can show restrictive physiology.16
Mitochondrial cardiomyopathiesMitochondrial cardiomyopathies are marked by abnormal myocardial structure and/or function, secondary to either a defective oxidative phosphorylation system, formed of subunits encoded by nuclear and mitochondrial DNA, or a defect in proteins essential for the replication, transcription, and maintenance of mitochondrial DNA, which are encoded by nuclear DNA, leading to mitochondrial DNA depletion or multiple mitochondrial DNA deletions.
The most common forms of mitochondrial cardiomyopathy are HCM, DCM, and excessive LV trabeculation.17 Mitochondrial cardiomyopathies frequently begin with progressive diastolic dysfunction and heart failure with preserved ejection fraction, and later progress to LVH with systolic dysfunction. Other cardiac tissues may also be affected, leading to conduction disease, arrhythmias, valvular disease, systolic dysfunction, or heart failure. Although the heart, being a high-energy-demand tissue, is one of the main organs affected by mitochondrial dysfunction, most patients display a clinical syndrome with degenerative, neuromuscular, ophthalmological, gastroenterological, auditory, or endocrine manifestations.18
This wide phenotypic and genotypic heterogeneity makes both suspicion and diagnosis of mitochondrial disorders and cardiomyopathy extremely complex. A high degree of clinical suspicion is usually warranted, given the absence of pathognomonic cardiac findings. Therefore, mitochondrial dysfunction should be suspected if, in addition to cardiomyopathy, the patient presents with systemic findings such as hearing loss, renal dysfunction, diabetes, or peripheral myopathy, or if there is a maternal inheritance pattern or family history of mitochondrial disease.19
The definitive molecular diagnosis of these disorders is also challenging. Several diagnostic schemes have been proposed to improve the chance of finding the primary genetic defect. Traditionally, the diagnostic approach used to rely on excluding other common metabolic disorders, and on subsequent biochemical and histochemical analysis of affected tissue, particularly muscle.
However, this approach is expensive, invasive, time-consuming, and often not definitive, as many patients remain undiagnosed.20 More recently, advancements in high-throughput sequencing technologies have completely transformed the mitochondrial disease diagnosis process.21 The diagnostic algorithm now favors a genomics-first approach, followed by histochemical and biochemical testing of tissue biopsy only if functional data are necessary to confirm the pathogenicity of identified genetic variants. This genomics approach involves a pipeline designed to simultaneously identify variants of low-level heteroplasmic mitochondrial DNA and nuclear DNA. This method can also detect pathogenic variants in non-mitochondrial genes, enabling the diagnosis of phenocopies of mitochondrial disorders. However, some limitations remain, and the order of investigations needs to be contextual, depending on the clinical situation. Due to the phenomenon of heteroplasmy, some low mutant load heteroplasmic mitochondrial DNA variants in blood samples may be missed, necessitating the sequencing of muscle mitochondrial DNA in these cases. Additionally, low-level heteroplasmic variants may not always be clinically relevant, and nuclear mitochondrial DNA sequences (NUMTs) may also cause false positive and false negative results. In both cases, targeted variant analysis in clinically affected tissues (such as skeletal muscle) is crucial to determine whether a specific variant detected in the blood is the cause of disease in an individual.22 Other challenges mainly involve clinical interpretation of the functional significance of variants identified by whole-exome or whole-genome sequencing, which may require deep clinical phenotyping, functional studies, and family segregation studies.23
RASopathiesThis group comprises congenital multisystem disorders caused by pathogenic variants in genes encoding signal transducers and regulatory proteins functionally linked to the Ras/mitogen-activated protein kinase (MAPK) pathway. Collectively, these disorders have an estimated prevalence of 1 in 1000-1 in 2500 live births. RASopathies present autosomal dominant inheritance and include Noonan syndrome, Noonan syndrome with multiple lentigines, cardiofaciocutaneous syndrome, and Costello syndrome.24
The two most common syndromes associated with HCM are Noonan syndrome and Noonan syndrome with multiple lentigines, with around 25% of Noonan syndrome patients presenting HCM. Although each RASopathy exhibits a unique clinical phenotype, these syndromes share many overlapping characteristics which should function as clinical clues, including growth retardation, distinctive craniofacial features, cognitive impairment, renal malformations, bleeding disorders, variable predisposition to certain cancers, and congenital heart disease (CHD) (aortic and pulmonary valve stenosis, atrioventricular septal defect).25,26
After CHD, HCM is the second most common cardiovascular abnormality observed in RASopathies, with worse clinical outcomes when associated with early onset presentation, and a mean age at diagnosis of six months. Compared with sarcomeric HCM, HCM in RASopathies (R-HCM) is associated with a higher prevalence of congestive heart failure and shows increased prevalence and severity of LV outflow tract obstruction. This is associated with higher rates of hospitalizations for heart failure and need for septal myectomy during childhood.
These disorders exhibit significant genetic heterogeneity, with causative mutations found in various genes, including PTPN11, RAF1, BRAF, SOS1, KRAS, NRAS, RIT1, SHOC2, MEK1, and CBL. The main genes associated with HCM are RAF1 (65%), NRAS (39%), RIT1 (36%) and PTPN1 (20%).27
The combination of biventricular involvement alongside the previously mentioned phenotypic characteristics can raise suspicion of R-HCM; right ventricular outflow tract obstruction may also be present.24
In addressing the management of cardiovascular complications, the use of selective beta-blockers is recommended to ameliorate symptoms associated with biventricular obstruction. In more severe cases, surgical myectomy and orthotopic heart transplantation may become necessary.1 With regard to targeted therapies, researchers have pioneered the development of various drug classes focusing on inhibiting Ras activation and components of the MAPK or PI3K/AKT pathways. Notably, some of these drugs, which were initially approved for treating cancer, hold promise for potential application in the treatment of RASopathies.28
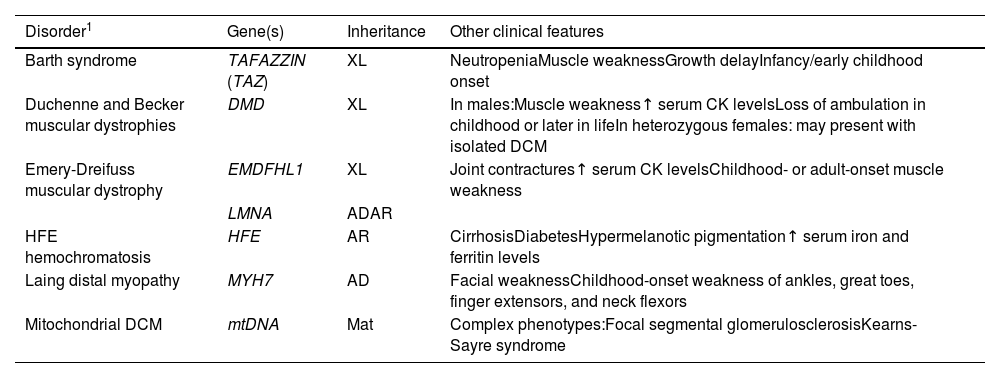
Syndromic dilated cardiomyopathyAs mentioned above, although in most cases genetic DCM primarily affects the heart, certain syndromic genetic conditions incorporate DCM as one of their features. Consequently, it is imperative to meticulously assess the family history and conduct a thorough physical examination to eliminate the possibility of syndromic disease. Among the multiple tissues that may be affected, skeletal muscle is the most often affected. Table 1 provides a comprehensive overview of the genes linked to these syndromic cardiomyopathies.
Genes associated with syndromic dilated cardiomyopathy.
| Disorder1 | Gene(s) | Inheritance | Other clinical features |
|---|---|---|---|
| Barth syndrome | TAFAZZIN (TAZ) | XL | NeutropeniaMuscle weaknessGrowth delayInfancy/early childhood onset |
| Duchenne and Becker muscular dystrophies | DMD | XL | In males:Muscle weakness↑ serum CK levelsLoss of ambulation in childhood or later in lifeIn heterozygous females: may present with isolated DCM |
| Emery-Dreifuss muscular dystrophy | EMDFHL1 | XL | Joint contractures↑ serum CK levelsChildhood- or adult-onset muscle weakness |
| LMNA | ADAR | ||
| HFE hemochromatosis | HFE | AR | CirrhosisDiabetesHypermelanotic pigmentation↑ serum iron and ferritin levels |
| Laing distal myopathy | MYH7 | AD | Facial weaknessChildhood-onset weakness of ankles, great toes, finger extensors, and neck flexors |
| Mitochondrial DCM | mtDNA | Mat | Complex phenotypes:Focal segmental glomerulosclerosisKearns-Sayre syndrome |
↑: increased; AD: autosomal dominant; AR: autosomal recessive; CK: creatine kinase; DCM: dilated cardiomyopathy; Mat: maternal inheritance; MOI: mode of inheritance; XL: X-linked.
Among the most common neuromuscular disorders with cardiac involvement are the dystrophinopathies. These disorders result from genetic variants in the DMD gene, which encodes the cytoskeletal structural protein dystrophin. This connects the sarcomeric proteins to the plasma membrane through the dystroglycan complex. Dystrophinopathies follow an X-linked pattern of inheritance and their clinical spectrum includes Duchenne muscular dystrophy (DMD), Becker muscular dystrophy (BMD) and X-linked DCM, as well as female DMD and BMD carriers.30 Their prevalence for all age groups is reported to be 1.12 per 10000 male individuals for DMD and 0.36 per 10000 male individuals for BMD. There is a higher predominance in Hispanic individuals.31
In the majority of cases along the spectrum, encompassing both DMD and BMD, patients initially exhibit skeletal muscle weakness. It is common for individuals to develop cardiomyopathy after the third decade of life; this may remain asymptomatic due to relative physical inactivity. Symptoms in female carriers can vary from mild weakness to raised creatine kinase and more severe manifestation. The incidence of cardiomyopathy in carriers increases with age, even in the absence of muscle symptoms.32 On the ECG, DMD patients often exhibit an increased R/S ratio in the right precordial leads, deep Q waves in the lateral leads, and conduction disease/arrhythmias. While echocardiography may offer limited usefulness in the early stages, CMRI can provide valuable insights for diagnosing and conducting cardiac screening in patients with established dystrophinopathies. Identification of subepicardial fibrosis in the inferolateral wall should arouse suspicion of DMD, of which this pattern is strongly characteristic, especially when encountered in cases of early-onset DCM accompanied by a family history of muscular dystrophy.33
Myotonic dystrophy type 1 (DM1) is the most common muscular dystrophy in the adult population. Its estimated prevalence is 5–20 per 100000 individuals.34 DM1 is a complex disorder affecting multiple body systems, including muscles (skeletal and smooth), the eyes, heart, endocrine system, and central nervous system. DM1 has three overlapping phenotypes: mild, classic, and congenital. The mild form presents with cataracts and mild myotonia and has a normal lifespan. Classic DM1 features muscle weakness, myotonia, cataracts, and often cardiac conduction issues, potentially leading to physical disability and reduced lifespan. Congenital DM1 exhibits severe weakness at birth, hypotonia, respiratory problems, and early mortality, often accompanied by intellectual disability. DM1 results from variants that lead to repeat expansion of a trinucleotide sequence (CTG) in the 3′-untranslated region of the DMPK (dystrophia myotonica protein kinase) gene on chromosome 19 at 19q13.3.35
Diagnosis relies on recognizing the characteristic muscle weakness and confirmation through molecular genetic testing. A CTG repeat length exceeding 34 is considered abnormal, with genetic testing detecting pathogenic variants in nearly all affected individuals. Mounting evidence suggests a link between cardiac manifestations and CTG expansion.35 The number of CTG repeats tends to impact the onset and severity of cardiac manifestations in DM1. These include conduction defects, which affect 40% of patients, often involving the His-Purkinje system and manifesting as minor abnormalities on the ECG such as PR, QRS, and HV interval prolongation; these may progress to severe defects with symptoms that can include syncope or sudden death. Tachyarrhythmias, such as atrial flutter and atrial fibrillation (up to 25% of patients), and ventricular arrhythmias (resulting from bundle branch re-entry, triggered activity, and fibrofatty degeneration) are common.36
Electrophysiology studies are essential for risk assessment and management, especially when patients exhibit symptoms (presyncope or syncope) or a family history of sudden death, given the potential for life-threatening arrhythmias. Invasive electrophysiological assessment should also be considered in patients with a PR interval ≥240 ms or QRS duration ≥120 ms or who are aged over 40 years and have supraventricular arrhythmias, or who are aged over 40 years and have significant LGE on CMRI.37
Friedreich's ataxiaFriedreich's ataxia (FRDA) has an estimated prevalence of ∼1/50000 live births. Its clinical manifestations include progressive ataxia of the limbs and trunk, dysarthria, scoliosis, bladder dysfunction, diabetes, and cardiac disease. Cardiomyopathy is present in approximately two-thirds of individuals with FRDA. The disease displays variable phenotypes, with early-onset cases showing more rapid progression and greater morbidity and mortality; they are typically wheelchair-bound by the age of 19–26 years, and mean mortality age is 39 years. By contrast, late-onset FRDA exhibits less severe cardiomyopathy and neurologic symptoms, with nearly average mortality.38
FRDA results from genetic variants affecting the mitochondrial protein frataxin due to GAA repeat expansions in the FXN gene (on chromosome 9 at 9q21.11). This impairs mitochondrial function, leading to energy production deficits and disrupted iron distribution. Diagnosis involves detecting biallelic pathogenic variants in FXN, with most cases having GAA repeat expansions.39 The size of the expansions correlates with the onset and severity of the disease, with longer expansions associated with higher mortality.40
HCM is present in about two-thirds of individuals with FRDA, and can advance to hypokinetic end-stage disease. This progression leads to LV dysfunction and a gradual reduction in wall thickness. Additional cardiac manifestations include supraventricular arrhythmias, notably atrial fibrillation. Features such as reduced LV ejection fraction, decreased wall thickness, T-wave inversion, fibrosis on CMRI, and elevated high-sensitivity troponin T are suggested as negative prognostic indicators.41
LaminopathiesLaminopathies are another noteworthy group of disorders, since they are associated with a broad range of phenotypes, some with neuromuscular and cardiac manifestations. They are caused by variants in the LMNA gene, which encodes the intermediate filament nuclear envelope proteins lamins A and C. Emery-Dreifuss muscular dystrophy (EDMD) is characterized by progressive wasting of skeletal muscles in the shoulder girdle and distal leg muscles, but its cardiac manifestations (such as DCM, AV block, and arrhythmias) may be similar to those in diseases associated with LMNA variants in which a cardiac phenotype predominates. On the other hand, the cardiac phenotype may also be driven by the location of the variant within the LMNA gene.42,43 The prevalence of EDMD is estimated to be 1.3–2 per 100000.44
In general, when addressing the cardiac aspects of neuromuscular disorders, particular attention should be paid to arrhythmias, necessitating close monitoring of electrocardiographic changes. While patients with neuromuscular diseases who have survived cardiac arrest or exhibit ventricular dysfunction are generally managed similarly to those without extracardiac manifestations, the decision to place an implantable cardioverter-defibrillator (ICD) should be carefully weighed, taking into account the overall prognosis, particularly in certain subtypes like Duchenne dystrophy. Conversely, some subtypes of neuromuscular disorders may justify a lower threshold for ICD implantation. For instance, individuals with limb-girdle type 1B or Emery-Dreifuss muscular dystrophy, along with indications for pacing, should be considered for an ICD. In conclusion, individuals within this population require a personalized approach when determining whether to proceed with pacemaker or ICD implantation.37
Transthyretin cardiac amyloidosisCharacterized by the deposition of insoluble amyloid fibrils, often resulting from destabilized transthyretin (TTR) tetramers, amyloidosis profoundly impacts multiple organs, particularly the heart. The accumulation of amyloid deposits within cardiac tissue precipitates a cascade of detrimental effects, encompassing oxidative stress, mitochondrial impairment, increased cardiac wall thickness, and diastolic dysfunction. These, in turn, culminate in a spectrum of severe cardiovascular complications, including congestive heart failure and arrhythmias.45
Several key red flags aid in the diagnosis and recognition of transthyretin cardiac amyloidosis (ATTR-CA). One notable indicator is a decrease in QRS voltages, often occurring before a significant increase in LV wall thickness becomes evident. Additionally, markedly elevated levels of natriuretic peptides and troponin are common in ATTR-CA patients.46 Echocardiography frequently reveals biatrial dilatation, increased LV and right ventricular wall thickness, and either normal or small LV cavity size. In some cases, pericardial effusion may also be present. The myocardium may display a granular sparkling appearance due to increased echogenicity from amyloid protein deposition, which contributes to the mismatch between QRS voltages and the degree of LV hypertrophy.47 For characterization of myocardial tissue with CMRI, the typical amyloid pattern is global subendocardial LGE, combined with changes in myocardial nulling kinetics, and increased T1 on TI mapping and extracellular volume.48
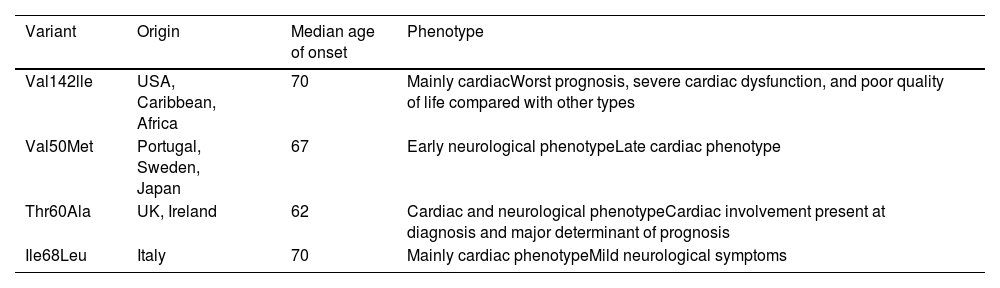
ATTR-CA can have two distinct causes: wild-type amyloid deposition, which occurs due to age-related misfolding of TTR, and hereditary, arising from autosomal dominant variants in the TTR gene. There are over 140 TTR variants spread across different geographical regions and giving rise to different phenotypic characteristics.49 These phenotypes encompass primarily cardiac, neuropathic, or combined manifestations, the latter with both cardiac and neuropathic symptoms (Table 2). While the specific variant strongly influences the resulting phenotype, nuanced variability may persist even among individuals harboring the same variant but from different regions.50
Genes associated with transthyretin cardiac amyloidosis.
| Variant | Origin | Median age of onset | Phenotype |
|---|---|---|---|
| Val142lle | USA, Caribbean, Africa | 70 | Mainly cardiacWorst prognosis, severe cardiac dysfunction, and poor quality of life compared with other types |
| Val50Met | Portugal, Sweden, Japan | 67 | Early neurological phenotypeLate cardiac phenotype |
| Thr60Ala | UK, Ireland | 62 | Cardiac and neurological phenotypeCardiac involvement present at diagnosis and major determinant of prognosis |
| Ile68Leu | Italy | 70 | Mainly cardiac phenotypeMild neurological symptoms |
One of the best-known TTR variants worldwide linked to a cardiac phenotype is Val50Met. This variant, prevalent across multiple continents including Europe, Asia, and parts of Africa and South America, demonstrates diverse manifestations based on geographic distribution. Endemic regions such as Portugal, Brazil, and Japan are typically characterized by an early-onset neuropathic phenotype with a positive family history, reflecting its autosomal dominant inheritance. By contrast, non-endemic areas present late-onset cardiac phenotypes, disproportionately affecting males.51
Hereditary transthyretin amyloidosis (ATTR) does not exhibit 100% penetrance and symptoms may only manifest later in adulthood. Notably, Val50Met homozygotes can remain asymptomatic. In Portugal, the cumulative risk of disease among individuals with the Val50Met variant is estimated at 80% by age 50 and 91% by age 70. By contrast, Sweden and France show lower penetrance rates.49
The Val142lle variant, originating from West Africa, is the most common in the USA, affecting 3–4% of African-Americans. It is characterized by late-onset restrictive cardiomyopathy.50 The Leu111Met and Ile68Leu variants, mostly observed in Denmark and Italy, lead to severe cardiomyopathy that manifests at a relatively young age.51 In the UK and Ireland, Thr60Ala is the most common variant, affecting about 1% of the population of north-western Ireland.49
Understanding of the various phenotypes and of the inherent heterogeneity within the disease opens the door to early identification and prognostic stratification, and potentially serves as a future focus for targeted therapeutic strategies.
DiscussionRecognizing syndromic cardiomyopathies is essential for effectively managing patients with these conditions, given the availability of targeted therapies and the distinct risk of potentially fatal arrhythmic events or heart block.36
The initial phase of diagnosing these diseases involves identifying the constellation of defining symptoms, with a conclusive diagnosis made possible through contemporary genetic testing techniques.5
Among syndromic cardiomyopathies presenting as HCM, severe hypertrophy is commonly observed in lysosomal storage diseases, with variable accompanying symptoms, depending on the specific type of disease.4
In the context of DCM, recognizing a neuromuscular disease as the cause is crucial, as these patients face a higher risk of fatal arrhythmias, necessitating specific risk stratification for sudden cardiac death.
Mitochondrial cardiomyopathies require a high degree of clinical suspicion, and their definitive diagnosis is very challenging. A genomics-first approach is now recommended; however, there is always a possibility of false positive or false negative results. Further functional, familial, or genetic studies in specific tissues are frequently needed to clarify the clinical relevance of genetic findings, and it must be borne in mind that a negative result does not exclude a mitochondrial disease.
Next-generation sequencing (NGS) technologies have enabled a higher diagnostic rate for syndromic cardiomyopathies; nevertheless, there are still limitations, including the possibility of failure to cover all regions of interest or to detect triple-repeat expansions, such as those in the DMPK1 or FRDA genes. Deep phenotyping and a multidisciplinary approach are therefore still essential.21
Overall, we have highlighted the inseparable interaction between genetics and this group of rare cardiomyopathies. The cardiologist should be able to identify the typical phenotypic traits of these diseases; however, management within a multidisciplinary team that includes geneticists is crucial.
ConclusionThe evolution of genetic diagnostic techniques, particularly NGS, has greatly facilitated the diagnosis of some cardiomyopathies, thereby shortening the diagnostic journey for rare diseases. A molecular etiological diagnosis enables personalized follow-up, tailored therapeutic interventions, and an accurate prognosis.
Conflicts of interestThe authors have no conflicts of interest to declare.
