
Congenitally corrected transposition of the great arteries (ccTGA) is a rare congenital heart defect. There are different subgroups according to the location of the heart in the thorax, apical position and situs.
ObjectivesThe purpose of this study was to assess pediatric patients with situs inversus (SI) ccTGA (SI-ccTGA), a rare subgroup of this condition, in detail.
MethodsThe records of patients with SI-ccTGA followed between January 1, 2010 and January 1, 2019 in our clinic were analyzed retrospectively. Demographic features, associated cardiac defects, arrhythmias and follow-up data were recorded.
ResultsTwenty-one out of 120 ccTGA patients had SI. The median age was 30 months (4 days-18 years). There were hemodynamically significant associated lesions in 85.7% (n=18) of the patients. A large ventricular septal defect was found in 16 patients (76.2%), severe pulmonary stenosis in 11 (52.4%), pulmonary atresia in six (28.5%), and severe tricuspid regurgitation in two (9.5%). Eleven out of 21 patients had biventricular physiology and eight had single-ventricle physiology. Bidirectional cavopulmonary anastomosis followed by a hemi-Mustard-Rastelli operation were planned for the remaining two patients. Twelve out of 18 patients with associated defects (66.6%) were operated and surgery was planned for three more patients (16.6%). The remaining three patients were scheduled for clinical follow-up. Arrhythmias developed in two (9.5%) patients on follow-up; ablation was performed in one of them and pacemaker implantation followed by cardiac resynchronization therapy was performed in the other. Two patients died during follow-up, one after a central shunt operation and the other preoperatively due to pneumonia and sepsis.
ConclusionSI-ccTGA is not a mirror image of situs solitus ccTGA (SS-ccTGA) due to important anatomic and physiologic differences between them. SI-ccTGA patients have a lower risk of tricuspid valve regurgitation than SS-ccTGA patients. The timing of clinical presentation of these patients mainly depends on the type and severity of the associated lesions, as in all subtypes of ccTGA.
A transposição congénita corrigida é uma doença cardíaca congénita rara. Existem subgrupos diferentes conforme a localização do coração no tórax, a posição apical e o situs.
ObjetivosEstudar os doentes pediátricos com cc TGA e situs inversus (SI), (SI-ccTGA), um subgrupo raro desta patologia.
Doentes e métodosForam avaliados retrospetivamente os relatórios de doentes com SI-ccTGA seguidos entre 1 de janeiro de 2010 e 1 de janeiro de 2019 na nossa clínica. Foram registadas as características demográficas, os defeitos cardíacos associados, as arritmias e dados clínicos de seguimento.
ResultadosVinte e um de 120 doentes com ccTGA tinham SI. A idade média foi 30 meses (quatro dias – 18 anos). Havia lesões hemodinamicamente significativas associadas em 85,7% (n=18) dos doentes. Foi detetado defeito do septo ventricular (DSV) extenso em 16 doentes (76,2%), estenose pulmonar (EP) grave em 11 (52,4%), atresia pulmonar (AP) em seis (28,5%) e regurgitação tricúspide (RT) grave em (9,5%). Onze doentes tinham fisiologia biventricular e oito fisiologia univentricular; a cirurgia de anastomose cavopulmonar bidirecional seguida da operação de Hemi-Mustard+Rastelli foi planeada para os restantes dois doentes. Doze de 18 doentes com defeitos associados (66,6%) foram operados, tendo a cirurgia sido planeada para três outros doentes (16,6%). Os restantes três doentes mantiveram-se em seguimento médico. Dois doentes (9,5%) em follow-up desenvolveram arritmias. Num foi efetuada ablação tendo sido implantado um pacemaker seguido de CRT no outro. Dois doentes morreram durante o seguimento, o primeiro após uma intervenção de shunt central e o outro no período pré-operatório devido a pneumonia e a sepsis.
ConclusãoA SI-ccTGA não é uma imagem em espelho de situs solitus ccTGA (SS-ccTGA). Os doentes com SI-ccTGA têm um risco menor de insuficiência tricúspide quando comparados com os doentes com SS-ccTGA. O timing da apresentação clínica destes doentes depende sobretudo do tipo e da gravidade de lesões associadas tal como em todos os subtipos de ccTGA.
Baron Rokitansky, in his 1875 atlas, was the first to describe the entity that is now known as congenitally corrected transposition of the great arteries (ccTGA). It is a rare cardiac condition, with a reported incidence of 1/33 000 live births, accounting for approximately 0.05% of congenital cardiac defects.1,2
Patients with ccTGA may present as situs solitus or situs inversus. Most cases of ccTGA have situs solitus, but situs abnormalities are reported in 19-34% of patients in pediatric series. In situs solitus, the normal anatomic pattern (not inverted), the morphologic left atrium (LA) and right atrium (RA) are located on the left and right sides, respectively. Conversely, in situs inversus, the mirror-image of situs solitus, the morphologic right atrium is located on the left and the left atrium on the right. The inversus infundibulum lacks left-sided infundibular free-wall musculature and has well-developed right-sided infundibular free-wall musculature.3–5
Associated cardiac anomalies are common with ccTGA, being seen in 80% of cases. The most important common associations are ventricular septal defect (VSD), pulmonary stenosis (PS), pulmonary atresia (PA), Ebstein's anomaly of the tricuspid valve, morphologic tricuspid regurgitation (TR) and complete heart block.3–7
The timing of clinical presentation of a patient with ccTGA mainly depends on the type and severity of the associated lesions.2,4,5 This feature is common to all ccTGA patients, despite the different frequencies of situs inversus and situs solitus. The long-term outcome of patients with ccTGA varies depending on the severity of the associated malformations and the timing of and approach to palliative surgical repair.
Although many detailed reports have been published on situs inversus and solitus ccTGA patients in adults, the number of reports on situs inversus ccTGA (SI-ccTGA) patients in pediatric age groups is limited.7
Our aim was to assess the characteristics and follow-up results of pediatric patients with SI-ccTGA.
MethodsData on 21 pediatric patients with SI-ccTGA followed in a pediatric cardiology clinic between January 1, 2010 and January 1, 2019 were analyzed retrospectively.
The echocardiographic study was based on the sequential segmental approach used to diagnose congenital heart defects. The study identified situs, atrioventricular (AV) and ventriculoarterial (VA) connections, spatial position of the ventricles and interventricular septum, characteristics of the arterial vessels, ventricular size, other associated cardiac lesions, and the orientation of the aortic arch. The position of the inferior vena cava and the aorta with respect to the spine was used to distinguish situs solitus from situs inversus. In short- and long-axis subcostal scans of the abdomen, the aorta was seen on the left of the spine and the inferior vena cava on the right in situs solitus. Conversely, in situs inversus, the aorta was on the right of the spine and the inferior vena cava located on the left.
SI-ccTGA with a segmental anatomic situs set of inverted atria, D-loop (solitus) ventricles, and D-TGA (solitus TGA) (anterior rightward aorta) indicates a well-developed right-sided subaortic infundibular free wall plus absence of a left-sided subpulmonary infundibular free wall. The AV alignments are discordant and the VA alignments are discordant.4
Patients with SI-ccTGA with a large VSD, moderate or severe PS, PA with VSD, moderate or severe TR, or Ebstein-like anomaly of the tricuspid valve were considered as having significant associated lesions. Patients with SI-ccTGA without any of these conditions were considered as having no associated lesions.1,5,6
Patients with heterotaxy, double-inlet AV connection, superior-inferior ventricles, and criss-cross hearts were excluded from the study.
In three patients, the echocardiographic findings were compared with results of cardiac catheterization to confirm the diagnosis.
Demographic data and clinical features (including congestive heart failure indicated by the clinical assessment), electrocardiographic (12-lead electrocardiogram [ECG] and 24-hour Holter monitoring), echocardiographic (cardiac situs, VSD location and size, Ebstein-like tricuspid valve, severity and degree of TR, degree of PS, aortic regurgitation, right and left ventricular dysfunction), and angiographic findings were noted. Duration of follow-up, mortality, type of surgery, arrhythmias, atrioventricular block (AVB), pacemaker implantation, cardiac resynchronization therapy (CRT), and ablations in SI-ccTGA patients were also recorded.
The study protocol was approved by the local ethics committee and the study was conducted in accordance with the principles of the Declaration of Helsinki.
Statistical analysisSPSS for Windows version 15 (SPSS, Chicago, IL, USA) was used for the statistical analysis. Continuous variables were expressed as median (minimum-maximum) and categorical variables were expressed as percentages.
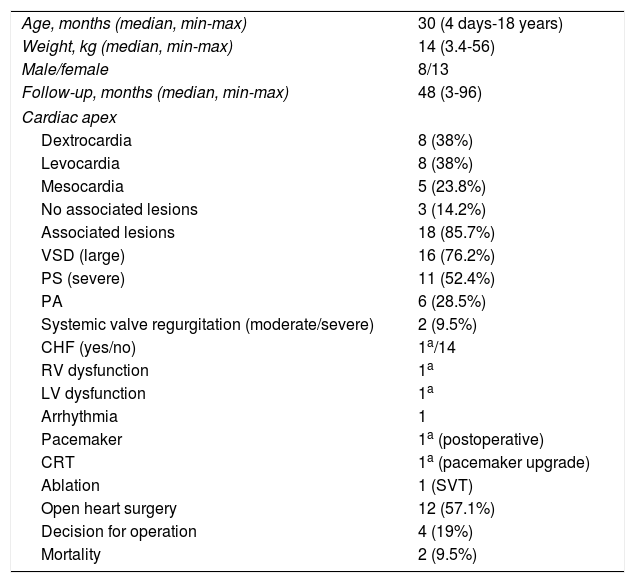
ResultsTwenty-one out of 120 ccTGA patients (17.5%) had SI-ccTGA. Eight patients (38%) were male and 13 patients (62%) were female. The median age at diagnosis was 30 months (4 days-18 years), and median follow-up was 48 months (3-96 months). The demographic and clinical characteristics of the study population are presented in Table 1.
General characteristics of the study population.
| Age, months (median, min-max) | 30 (4 days-18 years) |
| Weight, kg (median, min-max) | 14 (3.4-56) |
| Male/female | 8/13 |
| Follow-up, months (median, min-max) | 48 (3-96) |
| Cardiac apex | |
| Dextrocardia | 8 (38%) |
| Levocardia | 8 (38%) |
| Mesocardia | 5 (23.8%) |
| No associated lesions | 3 (14.2%) |
| Associated lesions | 18 (85.7%) |
| VSD (large) | 16 (76.2%) |
| PS (severe) | 11 (52.4%) |
| PA | 6 (28.5%) |
| Systemic valve regurgitation (moderate/severe) | 2 (9.5%) |
| CHF (yes/no) | 1a/14 |
| RV dysfunction | 1a |
| LV dysfunction | 1a |
| Arrhythmia | 1 |
| Pacemaker | 1a (postoperative) |
| CRT | 1a (pacemaker upgrade) |
| Ablation | 1 (SVT) |
| Open heart surgery | 12 (57.1%) |
| Decision for operation | 4 (19%) |
| Mortality | 2 (9.5%) |
CHF: congestive heart failure; CRT: cardiac resynchronization therapy; LV: left ventricular; PA: pulmonary atresia; PS: pulmonary stenosis; RV: right ventricular; SVT: supraventricular tachycardia; VSD: ventricular septal defect.
While cyanosis (n=12, 57%) and cyanosis with dyspnea (n=5, 24%) were the main symptoms on clinical assessment at presentation, 19% (n=4) of the patients were asymptomatic. Three out of four asymptomatic patients had no associated lesions and the other patient had systemic valvular regurgitation as the associated lesion. Nineteen patients had varying degrees of murmur on cardiac auscultation.
The apical position was dextrocardia in eight, levocardia in eight and mesocardia in five of the patients on chest X-ray, ECG and echocardiography. Cardiomegaly was only identified in one patient on chest X-ray. Electrocardiographic results of the patients with dextrocardia, levocardia and mesocardia, respectively, were as follows: QRS axis median +850 (min +510, max +2160), +800 (-800, +1120), +1200 (+1060, +1710); QRS duration 80 ms (40-176), 68 ms (56-90), 70 ms (60-86). Corrected QT interval (QTc) was 410 ms (380-432). Supraventricular tachycardia (SVT) (one patient) and ectopic atrial rhythm (three patients) were identified on ECG or 24-hour Holter monitoring at admission. There was no conduction type block on any ECG at admission.
When echocardiographic and angiographic records were analyzed, significant associated lesions were identified in 18 of the patients (85.7%). These were a large VSD in 16 patients (76.2%), severe PS in 11 (52.4%), and PA with VSD in six (28.5%). None of the patients had diminutive pulmonary arteries. Interestingly there was no Ebstein's anomaly in any of the patients and severe TR was only diagnosed in two patients (9.5%). The other three patients were considered to have SI-ccTGA without associated defects (14.2%).
Twelve out of 18 patients with associated defects (66.6%) were operated, while three patients with no associated defects were not. The median age of the operated patients was seven months (19 days-12 years). Eleven (52.3%) out of 21 patients had biventricular physiology and eight (38%) patients had single-ventricle physiology. Bidirectional cavopulmonary anastomosis (BCPA) followed by a hemi-Mustard-Rastelli operation were planned for the remaining two (9.5%) patients.
Eight out of 11 (52.3%) patients with biventricular physiology had associated lesions while the remaining three had none. Three (37.5%) of these eight patients with associated defects were operated; a Blalock-Taussig shunt was performed in two of these patients within one month of age with the diagnosis of VSD and PA. VSD closure with left ventricle-pulmonary artery conduit replacement was performed as a physiologic repair in one patient, and pulmonary banding was planned for two other patients (one with severe systemic valvular regurgitation and the other with moderate PS and systemic ventricular dysfunction). The other asymptomatic VSD-PS patients with no significant valvular regurgitation were scheduled for follow-up.
Four patients (three with PS and one with PA) with large VSD and left ventricular hypoplasia characterized by a small and hypertrophic ventricular chamber (z score <-2) and dysplastic cardiac valves, two patients with very large VSD (21 mm and 25 mm) with inadequate septum and PA, and another two patients with very large VSD (19 mm and 24 mm) with inadequate septum and severe subvalvular-valvular PS were classified as having univentricular physiology. The pulmonary gradients of patients with univentricular physiology and PS were between 80 and 110 mmHg.
BCPA was performed in nine (42.8%) out of the 12 operated patients. The Fontan operation was completed in a patient who previously underwent BCPA. A hemi-Mustard-Rastelli operation was planned for two patients following BCPA (age <10 years, right ventricular ejection fraction [EF] <40% and severe TR).
Due to the high operational workload of the cardiac center, four previously planned operations could not be performed during the study period. These operations were pulmonary banding (two patients), BCPA (one patient) and double switch (one patient).
As patients with no associated lesions did not have systemic valvular regurgitation, surgery was not performed in these patients.
On the baseline ECG of a patient with SI-ccTGA dextrocardia and SVT, the QRS axis was +1720, PR duration 145 ms, QRS duration 80 ms and QTc 390 ms. Successful radiofrequency ablation of a ‘concealed’ right posterolateral accessory pathway was performed in a patient on follow-up due to SVT; no ventricular dysfunction was identified during follow-up.
A single-chamber epicardial pacemaker was implanted in an operated SI-ccTGA patient (VSD closure plus morphologic left ventricle-pulmonary artery conduit) with postoperative AVB. A CRT device was implanted in one patient nine years after permanent pacemaker implantation due to biventricular dysfunction resulting from pacing-related asynchrony. The patient's EF increased from 20% to 45% and New York Heart Association (NYHA) functional class improved from IV to II.
Two patients (9.5%) died during follow-up. One was a 19-day old newborn who had a central shunt operation and died on the fifth postoperative day. The other was a one-year-old infant with associated VSD and PS lesions who died preoperatively due to pneumonia and sepsis.
DiscussionThe number of studies on adult SI-ccTGA patients in the literature is limited, and to the best of our knowledge this study is the first to assess children with SI-ccTGA, a rare congenital cardiac defect with varied clinical and treatment options. The results of the study can be summarized as follows.
(i) This anomaly should not be regarded as a mirror image of SS-ccTGA because of the important anatomic and physiologic differences between the two types of atrial arrangements.
(ii) SI-ccTGA patients have a lower risk of spontaneous AVB and TR than SS-ccTGA patients.
(iii) The timing of clinical presentation of the patient with ccTGA mainly depends on the type and severity of the associated lesions.
Different associated lesions may be seen in SS-ccTGA and SI-ccTGA patients.1,5 In one study, the incidence of VSD and PS was reported as 80% and 50% in SS-ccTGA patients and 70% and 40% in SI-ccTGA patients, respectively, without statistical difference.6 These figures are similar to those in our population.
In previous studies, tricuspid valve anomalies were reported in up to 91% of ccTGA patients, including thin and frail chordae tendineae and Ebstein-like anomaly.8–10 By contrast, Westerman et al. first noted the low prevalence of morphologically tricuspid valve dysfunction in ccTGA with situs inversus.11 They hypothesized that malalignment of the atrial and ventricular septum could also be the cause of the abnormal implantation of the tricuspid valve and that good septal alignment in patients with situs inversus might preclude not only AVB but also tricuspid Ebstein-like anomaly.11 Thus, in the absence of AV conduction disorders and tricuspid valve abnormalities, the long-term outcome of patients with this condition is likely better.
Oliver et al.6 did not report any Ebstein-like anomaly in their study of adult SI-ccTGA patients. Similarly there was no Ebstein-like anomaly in any of the 21 patients in the present study. Despite the low incidence of tricuspid valve anomalies, there was moderate or severe TR in two of the patients, which was isolated in one, while the other with TR had a CRT upgrade after pacemaker implantation due to postoperative complete AVB.
Systemic ventricular dysfunction before the age of 45 years has been reported in 50% of ccTGA patients with associated lesions and one third of those with no associated defects.12–14 Clinically significant regurgitation of the morphologic tricuspid valve is the major risk factor for ventricular dysfunction and poor long-term outcome.15–17 In addition, complete AVB itself or pacing-related asynchrony can also cause systemic ventricular dysfunction.18 One of our patients was upgraded to CRT due to biventricular dysfunction and congestive heart failure that developed some time after pacemaker implantation which led to severe pacing-related asynchrony. This patient's EF and NYHA functional class subsequently improved. Ventricular dysfunction did not develop in any other patient. This may be due to patients’ younger age and shorter follow-up durations.
About one-tenth of patients born with ccTGA have complete AVB.19 In patients born with normal cardiac conduction, the risk of developing AVB increases by 2% per year, until it reaches a prevalence of 30% in adulthood.20 In the study by Oliver et al.,6 30 SS-ccTGA and eight SI-ccTGA adult patients were assessed. In two-thirds of the 17 SS-ccTGA patients (57%) with complete AVB, this was found to be unrelated to surgery. The sole case of complete AVB detected among SI-ccTGA patients was shown to be surgery-related.
In a previous study at our clinic by Kasar et al.21 assessing 65 ccTGA patients for rhythm disturbances, 16 patients (24.6%) with complete AVB received implantable pacemakers. Nine of these patients (13.8%), all with SS-ccTGA, were found to have spontaneous AVB. In the present study there was only one case of AVB detected in SI-ccTGA patients, which was related to surgery; no spontaneous AVB was recorded.
Malalignment of ventricular and atrial septa and AV node abnormalities are reported to be the main reasons for heart block in patients with SS-ccTGA.22–24 The penetrating bundle that courses along the anterior segment of the septum in patients with SS-ccTGA courses on the posterior rim of the septum in SI-ccTGA patients, similarly to AV concordance.25–27 This could explain the normal AV conduction in our patients.
It has also been postulated that the posterior AV node connection could be related to the good septal alignment in these cases.24–28 Hosseinpour et al.24 suggested that association with PS or PA could explain the observed good septal alignment in SS-ccTGA, as was reported previously in SS-ccTGA.
However, this hypothesis was not supported by the adult study by Oliver et al.6 In six out of eight SI-ccTGA patients without spontaneous AVB, the pulmonary outflow tract was normal or dilated. In the present study, in contrast to the findings of Oliver et al.,6 17 out of 21 patients with SI-ccTGA (80.9%) had PS (n=11) or PA (n=6), and there was no known spontaneous AVB in any of the 21 patients. Congenitally corrected transposition by itself was not a direct indication for surgery. As in SS-ccTGA, the main determining factor for surgical treatment was additional cardiac conditions.2–4,7,12 In Oliver et al.’s study,6 surgery was performed in only two out of eight patients with SI-ccTGA, whereas 12 out of 21 patients were operated in our study (57.1%). The mean age was 40±17 years in Oliver et al. and 30 months (4 days-18 years) in the present study. The low surgical rate in Oliver et al. could be explained by the loss of patients with SI-ccTGA pre- or postoperatively before adulthood. Finally, unlike in the adult reports in the literature, timing for the operation was related to the associated defects, and the frequency and timing of the surgeries were similar to patients with SS-ccTGA.2,6,7,12
Study limitationsThis study is limited by its retrospective nature. Also, as our center is a relatively new pediatric cardiac surgery clinic, the young age of the patients and short follow-up periods were other limitations.
ConclusionIn conclusion, compared to previous reports, the incidence of spontaneous AVB and Ebstein-like tricuspid valve is lower in SI-ccTGA than in SS-ccTGA patients. Accordingly a lower rate of development of ventricular dysfunction in SI-ccTGA patients might be expected. More detailed studies with larger groups of pediatric patients are needed on this subject.
Authors’ contributionsTK: contributed to the conception or design of the work, contributed to drafting the work, provided final approval of the version to be published, agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
EO: revised the work, provided final approval of the version to be published, agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
PA: conception or design of the work, drafting the work, provided final approval of the version to be published, agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
YE: contributed to acquisition and analysis of data for the work, provided final approval of the version to be published, agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
AG: contributed to the conception or design of the work, revised the work, provided final approval of the version to be published, agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Conflicts of interestThe authors have no conflicts of interest to declare.
