










Dyslipidemia is associated with increased risk of cardiovascular disease and atherosclerosis, and hence with high morbidity and mortality. This study investigated the effects of the nitroxide 4-hydroxy-2,2,6,6-tetramethylpiperidine 1-oxyl (Tempol) on lipid profile and cardiac morphology in low-density lipoprotein (LDL) receptor gene knockout (LDLr-/-) mice.
MethodsMale LDLr-/- mice (three months old, approximately 22 g weight) were divided into the following groups: controls, including (1) standard chow (SC, n=8) and (2) high-fat diet (HFD, n=8); and treatment, including (3) standard chow + Tempol (SC+T, n=8) (30 mg/kg administered by gavage, once daily) and (4) high-fat diet + Tempol (HFD+T, n=8) (30 mg/kg). After 30 days of the diet/treatment, whole blood was collected for analysis of biochemical parameters (total cholesterol, triglycerides [TG], high-density lipoprotein [HDL], LDL, and very low-density lipoprotein [VLDL]). The heart was removed through thoracotomy and histological analysis of the left ventricle was performed.
ResultsA significant increase in TG, LDL, and VLDL and marked left ventricular hypertrophy (LVH) were demonstrated in the HFD group relative to the SC group (p<0.05), while Tempol treatment (HFD+T group) significantly (p<0.05) prevented increases in the levels of these lipid profile markers and attenuated LVH compared with the HFD group.
ConclusionIn this study, Tempol showed potential for the prevention of events related to serious diseases of the cardiovascular system.
A dislipidemia está associada com aumento do risco para as doenças cardiovasculares e aterosclerose, refletindo na alta morbidade e mortalidade associadas. Este estudo investigou os efeitos do nitróxido 4-Hydroxy-2,2,6,6-tetramethylpiperidine 1-oxyl (tempol) sobre o perfil lipídico e a morfologia cardíaca em camundongos nocaute para o gene do receptor da lipoproteína de baixa densidade (LDLR KO ou LDL-/-).
MétodosCamundongos machos (três meses de idade, pesando aproximadamente 22 g) foram divididos nos seguintes grupos: grupos controlo: (1) ração padrão ([RP] n=8) = camundongos LDL-/- + dieta padrão; (2) dieta rica em lipídios ([DRL] n=8) = camundongos LDL-/- + DRL; e grupos tratados: (3) RP + tempol (RP + T, n=8) = camundongos LDL-/- + dieta padrão + tempol (30 mg/kg, administrado por gavagem, uma vez por dia); (4) DRL + tempol (DRL + T, n=8) = camundongos LDL-/- + DRL + tempol (30 mg/kg). Após 30 dias de dieta/tratamento, o sangue total foi obtido para análise dos parâmetros bioquímicos (colesterol total [CT], triglicerídeos [TG], HDL, LDL e VLDL) e, através de uma toracotomia, o coração foi removido e uma análise histológica do ventrículo esquerdo foi realizada.
ResultadosFoi demonstrado um aumento significativo dos níveis de TG, LDL e VLDL, bem como uma considerável hipertrofia ventricular esquerda (HVE), no grupo DRL em comparação com o grupo RP (p<0,05); o tratamento com tempol (grupo DRL + T) preveniu significativamente (p<0,05) o aumento nos níveis destes marcadores de perfil lipídico e atenuou a HVE, em comparação com o grupo DRL.
ConclusãoTempol apresentou potencial para a prevenção de eventos que podem levar a graves doenças do sistema cardiovascular.
Cardiovascular disease is the leading cause of morbidity and mortality and is responsible for approximately 30% of all deaths, claiming approximately 17 million lives per year worldwide in 2012.1–3 Furthermore, many studies have firmly established the relationship between cardiovascular disease and metabolic disorders and underlying conditions such as dyslipidemia, diabetes, and hypertension.4–8
Dyslipidemia and associated atherosclerotic/cardiovascular events can present with intense inflammation and increased production of reactive oxygen/nitrogen species (ROS/RNS) from mitochondrial oxidative stress and/or the NADPH oxidase complex, which can cause oxidative modification of LDL, thus amplifying the inflammatory potential (i.e., recruitment of phagocytes and activation of the neutrophil oxidase [Nox]-2 system) and proatherogenic events. Moreover, uncontrolled dyslipidemia can have serious consequences for the cardiovascular system, resulting in morphological changes (left ventricular hypertrophy [LVH]), dysfunction, and even heart failure.9–16
Regulation of lipid metabolism is an important target for therapeutic intervention in dyslipidemic processes to prevent or reduce the risk or severity of cardiovascular disease, and appropriate intervention can have an impact on its clinical course. However, due to the high cost, prolonged use, and especially the adverse effects associated with some lipid-lowering drugs, a drug to control dyslipidemia that presents fewer side effects and a better cost/benefit ratio is highly desirable.17–19
Studies have explored other compounds with antioxidant properties in the prevention of cardiovascular disease.10,20 Over the last few decades, nitroxides have been widely investigated because of their antioxidant capabilities.20–22 Among them, 4-hydroxy-2,2,6,6-tetramethylpiperidine 1-oxyl (Tempol) is a superoxide dismutase (SOD) mimetic that shows a good partition coefficient, interacts with a broad spectrum of oxidants produced in the human body, and is able to break the chain of redox reactions.21,23,24
It has been shown that Tempol has radioprotective, chemopreventive, hypoglycemic, antihypertensive, antineoplastic, and cardioprotective effects, as well as protecting against ischemia-reperfusion injury. It also prevents obesity and neurodegenerative diseases and attenuates renal dysfunction and oxidative stress-induced injury. These biological effects derive at least partially from the ability of this nitroxide to scavenge ROS/RNS, as shown in several studies in which it has been proposed that Tempol can alleviate inflammatory diseases and reduce formation of extracellular traps (ETs) through its action on these oxidants.22,23,25 However, the beneficial anti-dyslipidemic effects of Tempol and its impact on cardiovascular events remain uncertain, and it is important to understand these actions by exploring different experimental animal models to find new therapeutic options. Therefore, the aim of this study was to assess the effects of Tempol on dyslipidemia and LVH in the well-established animal model of LDLr-/- mice.
MethodsEthics statementAll animal experiments were carried out in strict accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals (National Institutes of Health, Washington DC: The National Academy Press, 2011). This study was approved by the ethics committee on the use of animals (CEUA) of the University José do Rosário Vellano (UNIFENAS), approval number 01 A/2015.
Animals and experimental designIn this study, 40 three-month-old male mice, homozygous for the absence of the LDL receptor gene (LDLr-/-), background C57BL6, acquired from the Jackson Laboratory, Bar Harbor, Maine, USA and weighing approximately 22 g were used. These animals were supplied by the UNIFENAS breeding colony and were housed at a controlled temperature (25±1°C) in a light-controlled room with a 12-h light/dark cycle. After acclimation, the mice were randomly and equally divided into five experimental groups of eight animals per group (n=8), constituted as follows.
Control groupsThe standard chow (SC) group were fed a standard chow (Nuvital®, Nuvilab, Colombo, Brazil) for 30 days, and the high-fat diet (HFD) group were fed a high-fat diet (20% total fat, 1.25% cholesterol and 0.5% cholic acid; total 2.89 kcal/g; Instituto Tecnológico de Alimento, Campinas, SP, Brazil) for 30 days.
Treatment groupsThe standard chow + Tempol (SC+T) group were fed a standard diet (Nuvital®) and treated with Tempol (97.0%, Sigma-Aldrich, St. Louis, MO, USA) for 30 days at a dose of 30 mg/kg administered by gavage once daily, and the high-fat diet + Tempol (HFD+T) group were fed a high-fat diet (20% total fat, 1.25% cholesterol, and 0.5% cholic acid) and treated with Tempol for 30 days at a dose of 30 mg/kg administered by gavage once daily. Additionally, another group (HFD+S) were fed a high-fat diet (20% total fat, 1.25% cholesterol, and 0.5% cholic acid) and treated with simvastatin (Medley, SP, Brazil) at a dose of 20 mg/kg administered by gavage once daily. All animals were fed their respective diets and received water ad libitum.
Biological samplesAfter 30 days, mice were maintained on a fasting diet for 12 hours and then anesthetized by intramuscular injection of ketamine (40 mg/kg, Bayer AG and Parke-Davis®, Berlin – Bayer, Leverkusen, Germany) and xylazine (6 mg/kg, Bayer AG and Parke-Davis®, Berlin – Bayer, Leverkusen, Germany). Absence of the neuromuscular reflex was used to verify the anesthetic effect. Blood was collected via retro-orbital puncture (800 μl) using heparinized capillary tubes. After euthanasia and thoracotomy, 6 ml of 1.34 mM KCl was injected into the hearts through the left ventricle, and the organ was removed.26
Analysis of lipid profilesBiochemical markers were assessed by standard methods using commercially available kits (Labtest, MG, Brazil). Plasma levels of total cholesterol (TC, Liquiform kit) and fractions (triglycerides [TG, GPO-ANA kit], high-density lipoprotein [HDL, cholesterol HDL kit], and low-density lipoprotein [LDL, cholesterol LDL kit]) were determined by the endpoint colorimetric method (absorbance values readable spectrophotometrically at 500 or 540 nm). The level of very low-density lipoprotein (VLDL) was determined as previously described.27
Histological analysis of the heartBriefly, as previously described,26 the mouse hearts were dissected and the left ventricles were fixed in 10% neutral-buffered formalin for 48 h, and the fixed specimens were processed by a conventional paraffin-embedding technique for histological serial sections of 3-μm thickness. The serial sections were collected from the same plane, deposited on slides, and stained with hematoxylin and eosin for morphological analysis (Nikon optical microscope, TNB-04T-PL, magnification 40× or 100×). Measurement of left ventricular thickness followed standard criteria, using LGMC-image software, version 1.0. All histological analyses were performed by a single examiner using the double-blind method.
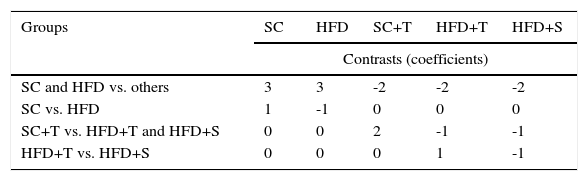
Statistical analysisThe effect of the intervention in the treatment groups was assessed with respect to TG, TC, VLDL, LDL, and HDL. Analysis of variance (ANOVA) was used to determine significant differences between the control and treated groups. Based on the results of the ANOVA/F test, orthogonal contrasts (comparisons) between the variables were performed, as shown in Figure 1.

Before analysis of the data by ANOVA, the Shapiro-Wilk test (α=5%) was performed. This showed that the data had a normal distribution under the null hypothesis of normality for all groups regarding the variables (p>0.05). For comparisons of body weight, lipid profile markers and LVH, the mean values ± standard error of the mean (SEM) or standard deviation (SD) of at least three experiments are shown, and the variables were analyzed by ANOVA, followed by Tukey's, Scott-Knott's, and Bonferroni's tests for multiple comparisons of the means (α=5%). Additionally, interactions between the groups and times (0 and 30 days) were considered, with body weight being assessed at 0 and 30 days. The main effects were also assessed separately for groups and times in terms of body weight. Sisvar (Lavras, MG, Brazil, 2008) and BioEstat 5.0 (Belém, Pará, Brazil, 2007) were used for the statistical analysis.
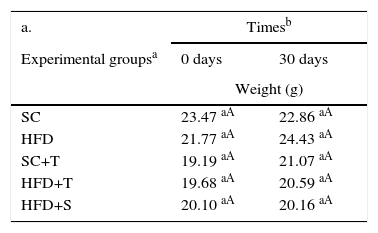
ResultsEffects of Tempol on the body weights of LDLr-/- miceResults of the comparisons of body weights according to the times at the beginning and end of the study period are shown in Table 1. No significant differences were observed between the groups over time (at 0 and after 30 days) (p>0.05) (Table 1, a). Table 1 also shows the overall mean weight (all groups) at 0 and 30 days, which was observed to increase after 30 days of the experiment (Table 1, b). In contrast, Table 2 shows that, in general, there was a decrease in mean body weight in the treated groups (SC+T, HFD+T, and HFD+S) compared to the control groups (SC and HFD vs. others) (p=0.000); however, there was no significant difference among the controls (SC vs. HFD). The treated groups did not differ statistically (p=0.886).
Means of body weights assessed in the study groups compared at 0 and 30 days.
| a. | Timesb | |
|---|---|---|
| Experimental groupsa | 0 days | 30 days |
| Weight (g) | ||
| SC | 23.47 aA | 22.86 aA |
| HFD | 21.77 aA | 24.43 aA |
| SC+T | 19.19 aA | 21.07 aA |
| HFD+T | 19.68 aA | 20.59 aA |
| HFD+S | 20.10 aA | 20.16 aA |
| b. Times (days) | Meansc |
|---|---|
| 0 | 20.44 a |
| 30 | 21.84 b |
HFD: high-fat diet; HFD+T: high-fat diet + Tempol; HFD+S: high-fat diet + simvastatin; SC: standard chow; SC+T: standard chow + Tempol.
p-values, estimates, and coefficients for the variables analyzed according to the orthogonal contrasts shown in Figure 1.
| Groups | SC | HFD | SC+T | HFD+T | HFD+S |
|---|---|---|---|---|---|
| Contrasts (coefficients) | |||||
| SC and HFD vs. others | 3 | 3 | -2 | -2 | -2 |
| SC vs. HFD | 1 | -1 | 0 | 0 | 0 |
| SC+T vs. HFD+T and HFD+S | 0 | 0 | 2 | -1 | -1 |
| HFD+T vs. HFD+S | 0 | 0 | 0 | 1 | -1 |
| Body weight | Total cholesterol | ||||
|---|---|---|---|---|---|
| Contrasts | Contrasts | ||||
| Groups | Estimates | p | Groups | Estimates | p |
| SC and HFD vs. others | 2.52 | 0.000b | SC and HFD vs. others | 131.83 | 0.000b |
| SC vs. HFD | -0.94 | 0.077c | SC vs. HFD | 157.42 | 0.001b |
| SC+T vs. HFD+T and HFD+S | -0.06 | 0.886c | SC+T vs. HFD+T and HFD+S | 73.90 | 0.058c |
| HFD+T vs. HFD+S | 0.00 | 0.992c | HFD+T vs. HFD+S | -25.25 | 0.297c |
| Triglycerides | LDL | ||||
|---|---|---|---|---|---|
| Contrasts | Contrasts | ||||
| Groups | Estimates | p | Groups | Estimates | p |
| SC and HFD vs. others | 164.40 | 0.000b | SC and HFD vs. others | 70.54 | 0.007b |
| SC vs. HFD | 162.87 | 0.016a | SC vs. HFD | 216.48 | 0.000b |
| SC+T vs. HFD+T and HFD+S | 144.48 | 0.014a | SC+T vs. HFD+T and HFD+S | 117.18 | 0.001b |
| HFD+T vs. HFD+S | -25.25 | 0.696c | HFD+T vs. HFD+S | -36.82 | 0.332c |
| VLDL | HDL | ||||
|---|---|---|---|---|---|
| Contrasts | Contrasts | ||||
| Groups | Estimates | p | Groups | Estimates | p |
| SC and HFD vs. others | 25.94 | 0.000b | SC and HFD vs. others | 0.077 | 0.990c |
| SC vs. HFD | 60.01 | 0.000b | SC vs. HFD | -22.75 | 0.221c |
| SC+T vs. HFD+T and HFD+S | 37.50 | 0.000b | SC+T vs. HFD+T and HFD+S | 5.01 | 0.535c |
| HFD+T vs. HFD+S | -18.21 | 0.083c | HFD+T vs. HFD+S | -25.25 | 0.957c |
| LVH | Contrasts | |
|---|---|---|
| Groups | Estimates | p |
| SC and HFD vs. others | 0.456 | 0.003b |
| SC vs. HFD | 0.812 | 0.001b |
| SC+T vs. HFD+T and HFD+S | 0.783 | 0.782c |
| HFD+T vs. HFD+S | 0.461 | 0.036a |
HDL: high-density lipoprotein; HFD: high-fat diet; HFD+T: high-fat diet + Tempol; HFD+S: high-fat diet + simvastatin LDL: low-density lipoprotein; LVH: left ventricular hypertrophy; SC: standard chow; SC+T: standard chow + Tempol.
Treatment with Tempol in the HFD+T group had no effect on TC levels (Figure 2), but prevented increases in plasma levels of TG, VLDL, and LDL compared with the control HFD group (Figures 3–5). Regarding the orthogonal contrasts assessing the intervention for the treated groups vs. control groups (all untreated groups or baseline groups), Table 2 shows that the treated groups (SC+T, HFD+T, and HFD+S) differed from the untreated groups (SC and HFD) (p=0.000) in terms of TC; however, this variable did not differ significantly when the groups were compared to each other (Figure 2). Table 2 also shows that TG levels fell in the treated groups compared with the untreated groups (p=0.000), a similar result to that presented in Figure 2. Regarding LDL (Table 2), the groups differed (p=0.000); the treated groups (SC+T, HFD+T, and HFD+S) presented lower mean LDL values than in the control groups (SC and HFD) (Table 2, Figure 4). No significant difference (p=0.332) was observed between the HFD+T and HFD+S groups (Figure 4). Table 2 shows that the treated groups (SC+T, HFD+T, and HFD+S) presented a decrease in VLDL compared to the control groups (SC and HFD) (p=0.000). There was no significant difference between the HFD+T and HFD+S groups (p=0.083) (Figure 5). No significant differences were observed in HDL between the treated and untreated groups (Table 2, p=0.1840) and among all groups (Figure 6).

Blood levels of total cholesterol in the experimental groups fed a standard diet and a high-fat diet. HFD: high-fat diet; HFD+T: high-fat diet + Tempol; HFD+S: high-fat diet + simvastatin; SC: standard chow; SC+T: standard chow + Tempol; TC: total cholesterol. Values are mean ± standard error of the mean; α=0.05; *p<0.05.

Blood levels of triglycerides in the experimental groups fed a standard diet and a high-fat diet. HFD: high-fat diet; HFD+T: high-fat diet + Tempol; HFD+S: high-fat diet + simvastatin; SC: standard chow; SC+T: standard chow + Tempol; TG: triglycerides. Values are mean ± standard error of the mean; α=0.05; *p<0.05.

Blood levels of low-density lipoprotein in the experimental groups fed a standard diet and a high-fat diet. HFD: high-fat diet; HFD+T: high-fat diet + Tempol; HFD+S: high-fat diet + simvastatin; LDL: low-density lipoprotein; SC: standard chow; SC+T: standard chow + Tempol. Values are mean ± standard error of the mean; α=0.05; *p<0.05.

Blood levels of very low-density lipoprotein in the experimental groups fed a standard diet and a high-fat diet. HFD: high-fat diet; HFD+T: high-fat diet + Tempol; HFD+S: high-fat diet + simvastatin; SC: standard chow; SC+T: standard chow + Tempol; VLDL: very low-density lipoprotein. Values are mean ± standard error of the mean; α=0.05; *p<0.05.

Blood levels of high-density lipoprotein in the experimental groups fed a standard diet and a high-fat diet. HFD: high-fat diet; HFD+T: high-fat diet + Tempol; HFD+S: high-fat diet + simvastatin; HDL: high-density lipoprotein; SC: standard chow; SC+T: standard chow + Tempol. Values are mean ± standard error of the mean; α=0.05; *p<0.05.
Regarding cardiac remodeling, representative histological images of the experimental groups are shown in Figure 6. The data show a correlation between morphology and left ventricular thickness. The mean thicknesses (in μm) in the five experimental groups are shown in Figure 7. There was no difference between the SC+T and SC groups (p=NS). Mean thickness was 0.6 μm less in the HFD+T group (p<0.05) than in the HFD group, and was 1.1 μm less in the HFD+S group (p<0.05) than in the HFD group and 0.5 μm less than in the HFD+T group. The difference between HFD+S and HFD+T was not significant (p>0.05) (Figure 8). Table 2 shows that, in general, the treated groups (SC+T, HFD+T, and HFD+S) presented less LVH than the control groups (SC and HFD) (p=0.003). Additionally, based on analyses between the individual groups (Figure 7), the control groups (baseline, SC and HFD) were significantly different from each other (p=0.001).
 and a high-fat diet (below). HFD: high-fat diet; HFD+T: high-fat diet + Tempol; SC: standard chow; SC+T: standard chow + Tempol; HFD+S: high-fat diet + simvastatin.")

In this study, we used a well-established animal model of LDL receptor knockout (LDLr-/-) mice, whose genetic background combined with a high-fat diet (environmental factor) is likely to lead to the development of severe dyslipidemia, and hence a high risk of cardiovascular disease. During the study, LDLr-/- mice were fed a standard or a high-fat diet for 30 days. Administration of Tempol (30 mg/kg once daily for 30 days) in mice on a high-fat diet showed a strong protective effect in controlling dyslipidemia and preventing damage to heart tissue (mitigating LVH).
LDLr-/- mice on a high-fat diet treated with Tempol showed a marked decrease in TG, LDL, and VLDL levels. Among the lipoproteins, LDL has been identified as one of the most important constituents of atheroma. As expected, LDLr-/- mice fed a high-fat diet presented a marked increase in LDL levels, and Tempol treatment decreased levels of this lipoprotein. Furthermore, increased TG levels have recently been shown to be associated with low HDL cholesterol levels, and hypertriglyceridemia is also one of the ‘deadly quartet,’ along with abdominal obesity, hypertension, and glucose intolerance.7,28,29
In our study, and in agreement with Kim et al.,11 LDLr-/- mice developed dyslipidemia with increases in TG, VLDL, and LDL (on a high-fat diet), and Tempol attenuated this condition; these results are similar to those found by Kim et al., in which Tempol treatment also prevented increases in these biomarkers of dyslipidemia.
Lipid disorders are among the most important risk factors for atherosclerotic cardiovascular disease, together with other chronic degenerative diseases with a prolonged natural history such as hypertension, obesity, and diabetes.4–7 These diseases have a complex relationship with one another; lifestyle and genetic inheritance, among other factors, are common to their etiologies. Treatment of dyslipidemia has the fundamental purpose of primary and secondary prevention of coronary artery disease, cerebrovascular disease, and peripheral arterial disease, and may also lead to the regression of xanthomas and reduce the risk of acute pancreatitis.30–32
Unlike LDL, HDL functions primarily in reverse cholesterol transport, reducing the formation of atherosclerotic plaque.33 In this study, HDL and TC levels in LDLr-/- mice did not differ significantly among the five groups evaluated. By contrast, the plasma HDL levels found by Kim et al.11 demonstrated that treatment with Tempol of apoE-/- mice maintained on a high-fat diet promoted increases in HDL levels, but these differences may be due to the characteristics of the different animal models used.34
Even on a normal diet, LDLr-/- mice may slowly develop dyslipidemia and atherosclerosis over time, and this process is accelerated with a high-fat diet.34 LDLr-/- mice develop moderate hypercholesterolemia (TC ∼250 mg/dl) when fed a standard diet and severe hypercholesterolemia when fed a high-fat diet. However, no differences between the groups were found regarding TC and HDL in this study; moreover, a closer relationship between these biomarkers was found. Furthermore, LDLr-/- mice normally become obese only when fed a high-fat diet with a fat content of more than 20%; thus, body weights differed only slightly among the groups, without statistical significance (Table 1) since only up to 20% total fat was used in this study.34
Simvastatin was used in this study as a control since this cholesterol-lowering agent is widely used preventively or as an adjuvant to correct lipid metabolism in patients with a predisposition to cardiovascular disease or primary hypercholesterolemia. Simvastatin is a prodrug that acts on the enzyme HMG-CoA reductase, preventing cholesterol synthesis and decreasing LDL, VLDL and TG.19,35 This statin, at a dose of 20 mg/kg, had significant effects on lipid profile markers and LVH, and its effects on TG, LDL, VLDL, and LVH were statistically similar to those caused by 30 mg/kg Tempol.
In LVH, increases in myocyte volume, coronary artery wall thickness, capillary rarefaction and extracellular fibrosis, and changes in energy metabolism, intracellular calcium, and myocardial contractility and relaxation are normally observed.13,14 LDLr-/- mice fed a high-fat diet for approximately 14 days develop atherosclerosis and an increased predisposition to cardiac damage, including LVH, with a mean 30% increase in cardiomyocyte diameter.26 In our study, morphological changes were observed in the untreated group of LDLr-/- mice fed a high-fat diet compared with those fed a standard diet, while treatment of these mice with Tempol attenuated these alterations, preventing LVH.
Regarding the underlying mechanisms, studies have shown that superoxide (O2-) production in LDLr-/- mice fed a high-fat diet is significantly higher than in LDLr-/- mice fed standard chow.36,37 Moreover, oxidative stress in these mice decreases nitric oxide (NO) and increases LDL oxidation. Thus, it is possible that antioxidant treatment reduces cardiac oxidative stress and prevents LVH by mitigating pathogenic mechanisms such as oxidative stress-mediated fibrosis.38
Ulasova et al. showed that quercetin, an antioxidant, can prevent LVH in ApoE-/- hypercholesterolemic mice,39 and other studies have shown that Tempol treatment can inhibit hypertension-induced oxidative stress-related LVH. In addition, Tempol (3 mmol) added to the drinking water of an experimental model of Dahl salt-sensitive rats for 10 weeks normalized LVH and reduced the cardiac expression of p22phox and Nox-2, mitochondrial uncoupling protein 2 and related oxidative stress. Thus, inhibition of cardiac ROS by Tempol prevented the cardiac fibrosis, remodeling and defective relaxation that underlie diastolic heart failure.40–42
The accumulated evidence suggests that Tempol can exert positive effects on dyslipidemia and prevent cardiac damage through multiple mechanisms; furthermore, a pleiotropic action is more plausible than a single mechanism in explaining the restoration of NO and nitric oxide synthetase (NOS) (NO is an important regulator of cardiac remodeling and is recognized as an anti-hypertrophic mediator).43,44
Furthermore, in this context, the action of Tempol in decreasing ROS is also relevant, since an increase in ROS in cardiomyocytes can activate the MAPK pathway, which has an important role in cardiac hypertrophy, and redox imbalance can also decrease the bioavailability of NO.43 Other ROS/RNS scavengers have been described as possible options for controlling atherosclerotic events and/or cardiovascular disease-related oxidative stress, since decreased antioxidant (i.e. SOD) activity and increased ROS generation have been described in such conditions, as in LVH.45–50
Several studies using other experimental models have reported the effects of Tempol on dyslipidemia, atherosclerotic events, and the cardiovascular system. The cardioprotective effect of Tempol demonstrated in this study using an LDLr-/- mice model is consistent with the findings of Zhu et al.,51 who demonstrated the action of Tempol (500 μM) on O2- in rat aortas, with a protective effect on the cardiovascular system by increasing the release of NO in the endothelium and causing vascular relaxation in aortas. Furthermore, the authors demonstrated that chronic treatment with Tempol (1 mM) added to the mice's drinking water restored the release of NO and aortic relaxation in rats.
Similarly to our findings, in an experimental model of rats with caerulein-induced pancreatitis, Marciniak et al.32 demonstrated that Tempol significantly decreased myocardial damage, mainly by attenuating oxidative stress-induced damage. Furthermore, as cited above, Kim et al.,11 using an experimental ApoE-/- mouse model, showed that Tempol (10 mg/g added to feed) can improve lipid profile, reduce the formation of pro-inflammatory cytokines and markers such as monocyte chemotactic protein and myeloperoxidase, helping reduce atherosclerotic plaque area and risk of myocardial damage.
Our study has potential limitations. Assessment of oxidative damage by measuring oxidative stress indices would have helped to demonstrate the role of oxidants in the events described herein. Measurement of other biochemical parameters such as blood glucose, interleukin-6, monocyte-chemotactic protein, myeloperoxidase, and serum amyloid A could also have supplemented these findings and established a stronger link between dyslipidemia, the genesis of atherosclerotic events, and subsequent cardiovascular complications.
Regarding the Tempol dose used in this study (30 mg/kg/day), other studies have reported the use of this nitroxide in animal models at doses of approximately 30 mg/kg or concentrations from 100 to 1000 μM/kg, once daily.11,32,51 These doses and concentrations do not display in-vivo toxicity (even doses of 300 mg/kg/day or 87 μM/kg have no toxic effects in vivo),23,52 are compatible with an EC50 for inhibiting the oxidative burst in vitro (50-400 μM),53 and concentrations up to 1000 μM appear to be safe, i.e., it does not show in-vitro pro-oxidant effects,54,55 which we consider may be a significant dose-limiting side effect in the systemic use of this nitroxide.
In addition to Tempol's previously mentioned possible side effects, caution should be exercised with nitroxides and other antioxidants because of the reductive stress that can occur with excessive use of this and other antioxidants, which may have short-, medium- and long-term effects. It has been reported that antioxidants influence the immune response (acting mainly by inhibiting the oxidative burst) and the onset of chronic diseases, such as cancer and cardiovascular disease, including cardiomyopathy.56
In summary, our study shows that the nitroxide Tempol is able to improve lipid profile and attenuate LVH in LDLr-/- mice fed a high-fat diet. These data suggest that this antioxidant can be a potent ally in preventing events related to cardiovascular disease.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that the procedures followed were in accordance with the regulations of the relevant clinical research ethics committee and with those of the Code of Ethics of the World Medical Association (Declaration of Helsinki).
Confidentiality of dataThe authors declare that no patient data appear in this article.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflicts of interestThe authors have no conflicts of interest to declare.
This research was supported by a grant from Fundação de Amparo à Pesquisa do Estado de Minas Gerais.
