







Portuguese WGMPD Recommendations and consensus documents on Transthyretin Amyloidotic Cardiomyopathy
Mais dadosEarly diagnosis of transthyretin amyloid cardiomyopathy (ATTR-CM) is crucial for better disease management and outcome. To ensure timely diagnosis, a multidisciplinary panel of Portuguese experts, including cardiologists, internal medicine specialists, and general practitioners, have developed a national consensus to aid physicians in enhancing the referral of patients with suspicion of ATTR-CM in Portugal.
A structured approach was used to develop the consensus: (1) an online survey aimed at identifying clinical red flags, patient journeys, and diagnostic tools related to ATTR-CM; (2) a face-to-face roundtable meeting where the survey findings were discussed and a consensus was reached on referral and diagnostic algorithms for ATTR-CM in Portugal; and (3) critical review of the proposed algorithms.
The referral and diagnostic algorithms for ATTR-CM in Portugal were developed considering current recommendations, but also the existence of a nationwide network of specialized cardiomyopathy clinics and national reference centers for familial amyloid polyneuropathy due to the endemic p.V50M variant.
This collaborative effort aims to enhance awareness, facilitate timely referrals and improve early diagnosis, ultimately ensuring better management of ATTR-CM patients in Portugal.
O diagnóstico precoce da cardiomiopatia por amiloidose de transtirretina (ATTR-CM) é fundamental para uma melhor gestão e prognóstico da doença. Para promover um diagnóstico atempado, um painel multidisciplinar de peritos portugueses, composto por cardiologistas, especialistas em medicina interna e médicos de medicina geral e familiar, desenvolveu um consenso nacional com o objetivo de apoiar os médicos no processo de referenciação de doentes com suspeita de ATTR-CM em Portugal.
Foi adotada uma abordagem estruturada para a elaboração do consenso: (1) um inquérito online direcionado para a identificação de sinais de alerta clínicos, trajetórias dos doentes e ferramentas de diagnóstico de ATTR-CM; (2) reunião presencial em formato de mesa-redonda, onde os resultados do inquérito foram discutidos e se alcançou consenso sobre os algoritmos de referenciação e diagnóstico de ATTR-CM em Portugal; e (3) revisão crítica dos algoritmos propostos.
Os algoritmos de referenciação e diagnóstico de ATTR-CM em Portugal foram desenvolvidos tendo em conta as recomendações internacionais atuais, bem como a existência de uma rede nacional de clínicas especializadas em miocardiopatias e centros de referência nacionais para a polineuropatia amiloidótica familiar devido à variante endémica p.V50M.
Este esforço colaborativo visa aumentar a sensibilização, facilitar a referenciação atempada e melhorar o diagnóstico precoce, assegurando, em última instância, uma melhor gestão dos doentes com ATTR-CM em Portugal.
Transthyretin amyloidosis (ATTR) is a disorder wherein misfolding, aggregation, and subsequent deposition of transthyretin (TTR) amyloid fibrils occurs extracellularly in various tissues, leading to organ dysfunction and diverse clinical phenotypes.1 Cardiac involvement and cardiomyopathy (CM), which occur due to fibril deposits in the myocardial interstitium, often result in a restrictive pathophysiology with severe cardiac diastolic dysfunction.1,2 Transthyretin amyloid cardiomyopathy (ATTR-CM) is a progressive and fatal condition if left untreated, resulting in progressive heart failure (HF) and increased risk of arrhythmias and conduction system disease, with patients experiencing a progressive decline in functional capacity and quality of life.1,2
The most common clinical presentation of ATTR-CM is left ventricular hypertrophy (LVH) and HF, manifesting with dyspnea, exercise intolerance, pulmonary congestion, fatigue, orthopnea, and edema.2,3 Other common cardiac manifestations of the disease include atrial fibrillation and conduction system disease, often needing pacemaker implantation.2,3 However, TTR deposition in other organs and tissues may lead to extracardiac manifestations, particularly in the musculoskeletal and peripheral and autonomic nervous systems.2,3 Musculoskeletal manifestations include carpal tunnel syndrome (CTS), spontaneous biceps tendon rupture, and lumbar spinal stenosis.2,3 Polyneuropathy primarily affects peripheral sensory and motor nerves. Damage to small myelinated and unmyelinated nerve fibers can result in sensory disturbances, including paresthesia, dysesthesia, allodynia, hyperalgesia and pain, typically starting in the feet. Over time, axonal degeneration progresses proximally, impairing light touch, vibration, and position sensation, contributing to motor deficits and walking difficulties and progressive sensory loss and motor weakness.2–4 Features of autonomic dysfunction include orthostatic hypotension, gastrointestinal manifestations, erectile dysfunction, and urinary manifestations.2–4
ATTR-CM can occur due to the deposition of amyloid fibrils of wild-type (wt) or variant (v) TTR protein (ATTRwt-CM and ATTRv-CM, respectively). In both cases, the protein is prone to misfold and aggregate, leading to organ deposits.2 ATTRwt-CM is associated with aging and is currently considered the most frequent form of cardiac amyloidosis worldwide.5 Mean age at diagnosis is 74 years.1 ATTRwt is predominantly characterized by cardiac involvement, whereas ATTRv has more heterogeneous phenotypes, depending on the TTR variant. Some variants cause a predominantly cardiac phenotype, while others lead to a predominantly neurologic phenotype, and some cause mixed phenotypes.1,6 Previously known as V30M,6 p.V50M6 is the most common TTR variant, particularly in Portugal, where it is endemic.7,8 The early-onset phenotype (age less than 50 years) described by Corino de Andrade typically starts with sensorimotor and autonomic dysfunction, evolving to motor impairment.9 By contrast, in the late-onset phenotype, motor impairment is often observed at the time of diagnosis and cardiac involvement is a frequent manifestation.8 The prevalence of this variant among Portuguese patients is remarkably high compared to other TTR variants, as evidenced by data from the Transthyretin Amyloidosis Outcomes Survey (THAOS), in which 99.1% of the enrolled Portuguese ATTRv patients harbored this variant.7,8 This highlights the epidemiological impact of this variant in the country and its impact on patient phenotypes.
The overall median survival from diagnosis in untreated patients is 3.6–4.8 years for ATTRwt-CM, 2.6 years for ATTRv-CM associated with the Val142Ile variant (a common variant in people of African descent), and 5.8 years for ATTRv-CM due to other TTR variants.6,10,11 Although ATTR-CM was deemed rare in the past, its diagnosis has increased worldwide in the past decade,12 but its exact prevalence is unknown.1 However, it is more prevalent in certain settings, affecting up to 18.8% of patients aged ≥50 years with hypertrophic cardiomyopathy,13 16.8% of patients aged ≥50 years with HF with preserved ejection fraction,14 and 16% of patients aged ≥65 years with severe aortic stenosis.15 Searching for ATTR-CM patients in these scenarios may help identify more cases.
ATTR-CM is often misdiagnosed, with patients being diagnosed several years after symptom onset.12 This is due to several factors, including (1) the overlap of ATTR-CM symptoms with other conditions; (2) the phenotypic heterogeneity of ATTR amyloidosis; (3) limited awareness and knowledge in the medical community about ATTR-CM; (4) the inaccurate perception of it being a rare condition; (5) the historical need for an invasive diagnosis; and (6) the lack, for many years, of disease-modifying therapy.3,16–18 One study found that the mean time to diagnosis was 6.1 years for ATTRwt-CM and 5.7 years for ATTRv-CM, with 34–57% of patients being previously misdiagnosed with other cardiac conditions.15 Misdiagnosis and late diagnosis may cause patients to undergo unnecessary exams and treatments, and, more importantly, hinders the timely initiation of disease-modifying treatment, which is crucial for preventing disease progression and improving patient outcomes.2,3 Without treatment, the disease progresses to a more advanced stage and patients develop more symptoms, present worse quality of life, and may need recurrent hospitalizations.3,17,18 Advanced disease stages also incur substantial healthcare costs, mainly from intensive inpatient care.19 As earlier treatment results in better outcomes,20 late diagnosis and treatment is associated with lower therapeutic benefit and, ultimately, disease-modifying treatment at a late stage of the disease (NYHA class IV) is no longer indicated. Ensuring early diagnosis and treatment has become even more important since the approval for ATTR-CM of tafamidis, a TTR stabilizer that decreases all-cause mortality, cardiovascular-related hospitalizations, and decline in functional capacity and quality of life in both forms of ATTR-CM.20,21 In addition, other therapies are on the horizon.22–26
In Portugal, a consortium of experts from various health institutions has produced a set of clear and concise recommendations to aid physicians in enhancing the referral of patients with suspicion of ATTR-CM. The key takeaways from their discussion are:
- 1.
Early detection of ATTR-CM is crucial and can be improved by raising physician awareness of red flags, fostering a high level of suspicion and prompt identification of the condition.
- 2.
Suspected or diagnosed ATTR-CM patients must be referred to a specialized CM clinic. CM experts can confirm the diagnosis and manage the condition, ideally in a multidisciplinary team setting.
- 3.
Physicians are advised to maintain a low threshold for suspicion and proceed with referrals even if only one or a few red flags are present.
This document was generated by bringing together a multidisciplinary panel of Portuguese clinicians, including cardiologists, internal medicine specialists, and general practitioners (GPs). These experts have comprehensive experience in cardiac amyloidosis and/or cardiomyopathies, advanced imaging, and HF. They have contributed scientifically to these fields and/or represent national centers of reference/excellence in managing patients with ATTR amyloidosis.
The project was developed in three stages. The first was completion of an online survey to gather input from the authors on high-value topics, such as red flags, patient journeys and diagnostic tools. The survey was designed to provide an overview of different national features: institutions, geographic regions and clinical settings. A survey of unmet needs regarding ATTR-CM in Portugal was also included. This first step aimed to help facilitate further discussion on the topics. The second step was a face-to-face roundtable meeting to discuss the survey results and to reach a consensus on simple and practical guidelines for the suspicion, diagnosis, and referral of ATTR-CM patients. The third step consisted of an iterative process of critical review of the content. This workflow was designed to optimize the collection of a broad range of perspectives, resolving differences in group opinions.
ResultsAll 12 authors replied to the survey prior to the first face-to-face roundtable meeting. The results can be found in the Supplementary Materials. Nearly all clinicians (92%) confirmed the existence of an organized CM clinic at their centers, reflecting their expertise in the field.
Suspicion of ATTR-CMSince diagnosis of ATTR-CM requires specific laboratory and imaging diagnostic tools beyond basic routine tests,2 a high level of clinical suspicion is essential.2 International guidelines recommend a system of red flags to raise the index of clinical suspicion for this condition, although there is no consensus, possibly due to different country contexts.27 The 2021 position statement of the European Society of Cardiology (ESC) Working Group on Myocardial and Pericardial Diseases is the most widely used guide among Portuguese clinicians for the diagnosis and treatment of cardiac amyloidosis.28 However, it remains unclear how many red flags should trigger a diagnostic work-up, whether some present higher predictive value than others, and how to combine them, suggesting an unmet need for a suspicion algorithm tailored to Portugal's national context.
The following red flags were deemed by the authors to be the most important, considering the situation in Portugal and their experience/expertise: unexplained LVH ≥12 mm, reduced global left ventricular (LV) longitudinal strain with apical sparing pattern, discordance between QRS voltage and LV wall thickness, late gadolinium enhancement with diffuse transmural or subendocardial pattern, increased signal on T1 mapping and/or extracellular volume (ECV), signs and symptoms of HF, newly developed intolerance to previously tolerated antihypertensive drugs, carpal tunnel syndrome (CTS), distal biceps tendon rupture, polyneuropathy, and orthostatic hypotension (Table 1 and online Supplementary Materials). These red flags have also been previously highlighted by other authors.2,3,5,27,28 Some were considered to have a greater predictive impact, prompting suspicion even when presenting alone, as is the case of unexplained LVH ≥12 mm and other suggestive findings from electrocardiography, echocardiography or cardiac magnetic resonance (CMR) such as global LV longitudinal strain with apical sparing pattern, discordance between QRS voltage and LV wall thickness, late gadolinium enhancement with diffuse transmural or subendocardial pattern, increased signal on T1 mapping and/or ECV (see online survey results, Supplementary Materials, and Table 2). Additionally, given the increasing known prevalence of ATTRwt-CM,12 physicians are encouraged to maintain a low threshold for suspicion, meaning that they should suspect ATTR-CM even in patients with one or few red flags. This is in line with a recent prospective observational study, which showed that most ATTR-CM patients with LVH ≥12 mm referred to a center of excellence presented three or fewer red flags.29
Cardiac and non-cardiac red flags for amyloid transthyretin amyloidosis.
| Cardiac | Non-cardiac |
|---|---|
| LVH (≥12 mm) | CTS |
| Reduced global longitudinal strain with apical sparing | Distal biceps tendon rupture |
| Diffuse transmural or subendocardial late gadolinium enhancement | Lumbar spinal stenosis |
| Increased signal on T1 mapping | Peripheral neuropathy |
| Increased ECV | Orthostatic hypotension |
| Discordance between QRS voltage and LV wall thickness | Intolerance to antihypertensive medications |
| Pseudo-infarction pattern | Renal abnormalities |
| Signs and symptoms of HF | Hepatomegaly |
| Severe aortic stenosis in the elderly | Gastrointestinal or genitourinary symptoms |
CTS: carpal tunnel syndrome; ECV: extracellular volume; HF: heart failure; LV: left ventricular; LVH: left ventricular hypertrophy.
Findings suggestive of transthyretin amyloid cardiomyopathy from different techniques.
| Technique | Suggestive findings3,28 |
|---|---|
| ECG | Discordance between QRS voltage and LV wall thicknessPseudo-infarction patternsConduction system defects (various degrees of atrioventricular block, fascicular block, intraventricular conduction delay, bundle branch block)Atrial fibrillation |
| Echocardiogram | Increased LV wall thickness (≥12 mm)Granular sparkling of the myocardiumDecreased global longitudinal strain with apical sparingDiastolic dysfunction grade 2 or worseDecreased mitral annular systolic velocity (<5 cm/s)Non-dilated left ventricleAV valve/right ventricular free wall/interatrial septal thickeningPericardial effusionBiatrial enlargement |
| CMR | Late gadolinium enhancement with diffuse or transmural subendocardial patternIncreased signal on T1 mapping and/or ECVAbnormal gadolinium kinetics |
| 99mTc-DPD/HMDP scintigraphy with SPECT | Perugini grade 2–3 (myocardial uptake of radiotracer)25 |
99mTc-DPD/HMDP: 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid/99mTc-hydroxymethylene diphosphonate; AV: atrioventricular; ECG: electrocardiogram; ECV: extracellular volume; CMR: cardiac magnetic resonance; SPECT: single-photon emission computed tomography.
Data from the online survey also indicated that Portuguese patients suspected of having ATTR-CM are most frequently referred by other medical specialties within the same institution, a scenario frequently seen in public hospitals and other public healthcare facilities. However, referrals also come from primary healthcare providers and private hospital settings (Supplementary Materials).
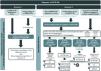
The suspicion algorithm proposed in Figure 1 reflects the national panorama and outlines four distinct scenarios in which ATTR-CM may be suspected, leading to a recommendation for the patient to be referred to a specialized CM clinic.

Algorithm for referring patients with suspicion of transthyretin amyloid cardiomyopathy to a specialized cardiomyopathy clinic. 99mTc-DPD/HMDP: 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid or 99mTc-hydroxymethylene diphosphonate; ATTR-CM: transthyretin amyloid cardiomyopathy; ATTRv: variant ATTR amyloidosis; BNPs: brain natriuretic peptides; CM: cardiomyopathy; CMR: cardiac magnetic resonance; CV: cardiovascular; ECG: electrocardiogram; HF: heart failure; LVH: left ventricular hypertrophy; NT-proBNP: N-terminal pro brain-type natriuretic peptide; TTE: transthoracic echocardiogram; TTR: transthyretin. aE.g., hypertension, diabetes, obesity47,48; bSee Table 2 for details; cLVH not explained by hypertension or valvular disease.
positive: Perugini grade 2–3 scintigraphy.31
The two most common scenarios encompass distinct clinical settings: centers with a specialized CM clinic and those without. These scenarios differ in their typical patient profiles, leading to differences in the most commonly observed red flags, as shown in Figure 1.
Centers without a specialized CM clinic have fewer resources and diagnostic tools, such as advanced imaging techniques, and their health professionals are less specialized in CM, often including those in primary healthcare settings. Although the complementary diagnostic tools to confirm a diagnosis are often unavailable, raising awareness and promoting early suspicion among GPs is essential, since they monitor many of the patients at risk for ATTR-CM. These centers will mainly refer patients with echocardiographic findings suggestive of ATTR-CM, such as unexplained LVH, to CM clinics.
Centers with a specialized CM clinic, which have access to advanced resources such as 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid or 99mTc-hydroxymethylene diphosphonate (99mTc-DPD/HMDP) scintigraphy, CMR and endomyocardial biopsies, will mainly refer to the CM clinic those patients with suspicion of ATTR-CM based on basic or advanced cardiac imaging, as well as patients with an established diagnosis of ATTR-CM.
Two less frequent but important scenarios were also considered sufficient to raise suspicion of ATTR-CM (Figure 1). The first encompasses patients who present a diagnosis of ATTR variant (ATTRv) amyloidosis due to non-p.V50M TTR variants, carriers of non-p.V50M TTR variants or individuals with a family history of ATTRv amyloidosis due to non-p.V50M TTR variants. The second consists of an incidental finding of grade 2 or 3 myocardial uptake of the radiotracer (99mTc-DPD/HMDP) on bone scintigraphy with planar and/or single-photon emission computed tomography (SPECT), performed for non-cardiac reasons.3,28
Importantly, unexplained LVH ≥12 mm was considered a red flag of high predictive value, which, alone or in conjunction with other red flags, should always be investigated and is therefore sufficient to trigger suspicion of ATTR-CM and referral of the patient to a specialized CM clinic (Figure 1). An LV thickness of ≥12 mm is the consensus threshold value in several guidelines,2,27,28 and is a cutoff above the gender-specific upper limit of normal. Women have lower LV wall thickness values than men, which may lead to underdiagnosis of ATTR-CM in women.30 LVH has the advantage of being detected by TTE, which is widely available.
Diagnosis of ATTR-CMAfter suspicion of ATTR-CM has been raised (Figure 1 and Table 2), the non-invasive diagnostic algorithm requires grade 2–3 99mTc-DPD/HMDP myocardial uptake in bone scintigraphy with SPECT, echocardiographic or CMR evidence of cardiac involvement by ATTR amyloidosis and exclusion of immunoglobulin light chain (AL) amyloidosis, in order to establish a diagnosis of ATTR-CM.
Exclusion of AL amyloidosis is mandatory, as these conditions have overlapping clinical, electrocardiographic and cardiac imaging features.2 The differential diagnosis may be performed by invasive and/or non-invasive diagnostic criteria.28
AL amyloidosis is excluded if all the following hematologic tests yield negative results, i.e., no monoclonal protein was detected: serum free light chain (FLC) assay; serum immunofixation electrophoresis (IFE); and urine IFE.3,28,31 The serum FLC assay measures the relative proportion of kappa and lambda light chains (κ/λ ratio), with monoclonality assumed by an abnormal ratio.27,28 Reference values for the κ/λ ratio depend on the estimated glomerular filtration rate (eGFR),32 as the κ/λ ratio increases with advancing chronic kidney disease due to the higher production of kappa light chains by the bone marrow and lower renal clearance of light chains in the presence of kidney disease. Reference values for the κ/λ ratio are depicted in Figure 2. Serum and urine IFE both measure the presence or absence of a monoclonal protein.2,3 Immunofixation must always be performed since electrophoresis alone lacks sensitivity.33 Hematology test results should be quickly performed and reviewed. This review should not be deferred due to the unavailability of scintigraphy results, as an abnormal light chain ratio or monoclonal protein detection should prompt a hematology consultation and potentially an endomyocardial biopsy to exclude AL cardiac amyloidosis (AL-CA).1–3

Algorithm for the diagnosis of transthyretin amyloid cardiomyopathy. AL: light chain amyloidosis; ATTR-CM: transthyretin amyloid cardiomyopathy; ATTRv: variant ATTR amyloidosis; CM: cardiomyopathy; CMR: cardiac magnetic resonance; eGFR: estimated glomerular filtration rate; FLC: free light chain; IHC: immunohistochemistry; LVH: left ventricular hypertrophy; MGUS: monoclonal gammopathy of undetermined significance; MS: mass spectrometry; TTE: transthoracic echocardiogram; TTR: transthyretin.
positive; negative; 99mTc-DPD/HMDP(99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid or 99mTc-hydroxymethylene diphosphonate) scintigraphy with single-photon emission computed tomography (SPECT): Perugini grade 2–331; biopsy: Congo red staining of amyloid deposits showing green birefringence when viewed under cross-polarized light28,39,40; aSee details in Table 2; $In the presence of the p.V50M variant, a biopsy is often required since 99mTc-DPD/HMDP scintigraphy can yield false negatives for this variant34–36; $Endomyocardial biopsy is the gold standard. An extracardiac biopsy may be considered and is helpful if positive, but the presence of a negative result should prompt further assessment through endomyocardial biopsy2,41; *Refined κ/λ FLC limits for normal ratio32; # Endomyocardial biopsy is the gold standard. An extracardiac biopsy may be considered and is helpful if positive, but the presence of a negative result should prompt further assessment through endomyocardial biopsy.2,41
Grade 2–3 99mTc-DPD/HMDP myocardial uptake in bone scintigraphy with SPECT and negative results on all three hematologic tests have demonstrated specificity and positive predictive values of 100% (confidence interval 98.0–100.0) for ATTR-CM in a large cohort.31 However, there are situations that can lead to false positives on 99mTc-DPD/HMDP bone scintigraphy31 (Table 3).
Possible false positives on 99mtechnetium 3,3-diphosphono-1,2-propanodicarboxylic acid or hydroxymethylene diphosphonate scintigraphy for detecting transthyretin amyloid cardiomyopathy.
| Condition | How to suspect | How to confirm |
|---|---|---|
| AL amyloidosis | Monoclonal protein tests | Histologic confirmation |
| AApoAI and AApoAII amyloidosis | Presence of concomitant kidney disease | Genetic testing |
| ApoAIV amyloidosis | Presence of concomitant kidney disease | Histologic confirmation |
| Aβ2M amyloidosis | Long-term dialysis (>9 years) | Histologic confirmation |
| Blood pool | Cardiac dysfunction can be present | Use SPECT for myocardial uptake detection; delay acquisition |
| Rib fractures, valvular/annular calcifications | Use SPECT for myocardial uptake detection | |
| Recent myocardial infarction (<4 weeks) | Clinical history | Use SPECT for diffuse myocardial uptake detection |
| Hydroxychloroquine cardiac toxicity | Clinical history | Histologic confirmation |
AApoAI: apolipoprotein AI amyloidosis; AApoAII: apolipoprotein AII amyloidosis; AApoAIV: apolipoprotein A-IV amyloidosis; Aβ2M: beta2-microglobulin amyloidosis; AL: light chain amyloidosis; SPECT: single-photon emission computed tomography.
The proposed diagnostic algorithm is based on the situation in Portugal, the authors’ expertise and experience, and current international guidelines (Figure 2). In Portugal, this algorithm is primarily implemented in national reference centers for familial amyloid polyneuropathy due to the p.V50M variant or in centers with specialized CM clinics.
Two possible initial scenarios were considered: (1) patients with a diagnosis of ATTRv amyloidosis due to the p.V50M variant, carriers of the p.V50M variant, or individuals with a family history of ATTRv amyloidosis due to the p.V50M variant and (2) patients with suspicion of ATTRv-CM due to other TTR variants or ATTRwt-CM.
In the first scenario, patients are referred to Portuguese reference centers for familial amyloid polyneuropathy due to the p.V50M variant. Cardiac imaging features suggestive of ATTR-CM would raise suspicion of the condition and a diagnosis of ATTR-CM could be confirmed through the non-invasive diagnostic algorithm or, if necessary, extracardiac or endomyocardial biopsy (Figure 2). In the case of the TTR variant being p.V50M, a biopsy is often required to confirm a diagnosis of ATTR-CM, since 99mTc-DPD/HMDP bone scintigraphy has low sensitivity and can yield false negatives for this variant, particularly in patients with early-onset disease.34–36
In the second scenario, patients are referred to specialized CM clinics, where the non-invasive diagnostic algorithm is applied, including the hematologic tests mentioned above along with 99mTc-DPD/HMDP scintigraphy with SPECT. Based on the results of these tests, there are four potential patient pathways that could be followed (Figure 2). Despite the differences in access to diagnostic tools, physicians working in centers without a specialized CM clinic (including those in primary healthcare settings) are encouraged to order all possible exams simultaneously with the patient's referral to a specialized CM clinic, if this helps to speed up diagnosis.
Distinguishing between different types of ATTR-CM by TTR genetic testing is important regardless of age, as about 5% of ATTR-CM patients aged ≥70 years (10% among females) have ATTRv.37 Genetic testing is also recommended for relatives of patients with ATTRv-CM, along with genetic counseling for patients and their families.27,28
Although they are unable to identify the type of amyloid, imaging techniques like CMR can provide helpful additional tissue characterization and high-resolution morphologic and functional assessment,38 and are thus recommended here as a complementary diagnostic method. Multimodality imaging techniques such as echocardiography, CMR, and radionuclide imaging are becoming more widely available. These non-invasive methods offer comprehensive cardiac imaging and could potentially be useful in monitoring amyloid burden and assessing its impact on cardiac function, for instance, in response to treatment.39
A biopsy positive for amyloid deposits has the pathognomonic histologic property of green birefringence when viewed under cross-polarized light after staining with Congo red.28,39,40 Although endomyocardial biopsy is highly specific, it is not widely available, requires technical expertise, and is an invasive procedure with potential risk for complications. Hence, cardiologists are often hesitant to proceed to biopsy in older adult patients or in those with early symptoms.39 Extracardiac biopsies, such as those taken from the abdominal fat pad, salivary glands, skin, peripheral nerve, kidneys, or bone marrow, are easier to perform.27,39,41 However, they have low sensitivity, and their yield in diagnosing ATTR amyloidosis is limited, particularly in the wild-type form.42 Extracardiac biopsies may be used as an alternative to endomyocardial biopsy, but given the high number of false negatives, further assessment is warranted in the presence of a negative result.2,41
Amyloid typing should be performed by immunohistochemistry (IHC) or mass spectrometry (MS) on a tissue biopsy by a trained clinical pathologist.28,39–41 IHC is a widely available technique, but it may lack specificity and sensitivity.40 MS is a more labor-intensive technique but has shown around 100% specificity and sensitivity in identifying the amyloid type, and should be used in cases of inconclusive IHC results.40,43 Reports have emerged of multiple myeloma patients with features of AL-CA (including abnormal serum FLC results), but ATTR-CM was in fact the confirmed diagnosis after biopsy and MS.44,45 Moreover, monoclonal gammopathy of undetermined significance (MGUS) has been estimated to affect 6.6% of people over the age of 80 years,46 which can lead to an increased risk of misdiagnosis in this population. Data from our online survey indicate that patients with abnormal findings suggestive of both ATTR-CM and AL-CA are the cases that give rise to the most uncertainty among experts (Supplementary Materials). Whenever this is the case, MS should be used for amyloid typing.
This algorithm shares several features with those from international societies and national consensus documents, namely: (1) the importance of differentiating between ATTR-CM and AL cardiac amyloidosis and the cornerstone techniques for doing so through a non-invasive diagnosis; (2) the possible diagnoses arising from different combinations of results (ATTR-CM, AL-CA, MGUS); and (3) the need for TTR genetic testing in patients with a definite diagnosis of ATTR-CM.2,3,5,27,28 Differences occur, for example, in the timings of the diagnostic techniques. The diagnostic algorithms of the World Heart Federation consensus, the ACC expert consensus, and the Amyloidosis Research Consortium expert consensus all suggest performing the hematologic tests first and, depending on their results, proceeding with 99mTc-DPD/HMDP bone scintigraphy or a biopsy,2,3,41 whereas in the present algorithm, hematologic tests and scintigraphy are recommended simultaneously. This diagnostic algorithm more closely resembles that of the position statement of the ESC Working Group on Myocardial and Pericardial Diseases.28 However, an important difference is that those guidelines rely more heavily on CMR results for diagnosis.28 Moreover, guidelines differ on when to opt for an extracardiac or an endomyocardial biopsy.2,3,5,27,28 Particularly in patients with a positive TTR genetic test and with no other cause for the cardiac phenotype, an extracardiac biopsy of an affected organ may be considered and is helpful if positive, but a negative result should prompt further assessment through endomyocardial biopsy.
Other considerationsImproving the early diagnosis of ATTR-CM requires a collaborative approach from a multidisciplinary team of physicians. The key factor is maintaining a high level of suspicion and ensuring timely referrals to specialized CM clinics, ideally with all necessary complementary exams pre-ordered to expedite and facilitate the diagnostic process. Full diagnosis and management of these patients should be carried out by CM specialists.2 These are uniquely positioned to determine whether the patient will meet the criteria for and benefit from the available disease-modifying therapeutic options (also preventing their misuse). This is in line with the results of the online survey, in which 83% of the authors mentioned the reason patients were referred to them as the “need to institute therapy,” particularly disease-modifying therapy (Supplementary Materials). In addition, CM specialists are uniquely equipped to manage these patients beyond these therapies. For instance, patients whose HF etiology is ATTR-CM must have their conventional prognosis-modifying therapy for HF (e.g., beta-blockers, angiotensin-converting enzyme inhibitors) reviewed and adjusted2 since some of these drugs may be poorly tolerated or ineffective2 in these patients.
ConclusionsA group of Portuguese experts on cardiomyopathies sought to overcome the lack of national recommendations regarding ATTR-CM by providing strategies to support any physician who might cross paths with these patients in their clinical practice. This work focuses on increasing awareness among physicians about the red flags for ATTR-CM, encouraging them to maintain a low threshold for suspicion, in order to achieve timely diagnosis and referral. This work seeks to empower Portuguese physicians with the knowledge to:
- •
identify patients with ATTR-CM by understanding how to suspect it, how to recognize clinical red flags, and how to prioritize them according to available resources;
- •
complete the diagnosis by knowing which diagnostic tools can be used in different settings and interpreting their results accurately; and
- •
appropriately refer these patients to a specialized CM clinic where they can receive a complete diagnosis and appropriate management.
This article is part of a supplement entitled ‘Portuguese WGMPD Recommendations and consensus documents on Transthyretin Amyloidotic Cardiomyopathy’ which is sponsored by Grupo de Estudo de Doenças do Miocárdio e do Pericárdio da Sociedade Portuguesa de Cardiologia - Portuguese Society of Cardiology Working Group on Myocardial and Pericardial Diseases.
This project, including the online survey and face-to-face roundtable meeting, was sponsored by Pfizer (EM-PRT-ARC-0037). Medical writing support was provided by Q2Science and was funded by Pfizer. The contents of this article are the exclusive responsibility of its authors.
The following are the supplementary data to this article:

