




It is unclear whether left ventricular noncompaction (LVNC) is a distinct cardiomyopathy or a morphologic manifestation of different cardiomyopathies. We previously reported a case of LVNC in a Fabry disease (FD) patient, but it remains to be clarified whether LVNC is a cardiac manifestation of FD, a coincidental finding or an overdiagnosis, which has major therapeutic implications. This study aims to determine the prevalence of FD among patients with LVNC.
MethodsWe performed a retrospective study including all patients diagnosed with LVNC in eight hospital centers. Diagnosis of LVNC was based on at least one echocardiographic or cardiac magnetic resonance criterion. FD screening was performed by combined enzyme and genetic testing.
ResultsThe study included 78 patients diagnosed with LVNC based on the Jenni (84.6%), Stöllberger (46.2%), Chin (21.8%), Petersen (83.8%) and Jacquier (16.2%) criteria. Left ventricular systolic dysfunction was present in 48.7%. Heart failure was found in 60.3%, ventricular dysrhythmias in 21.6% and embolic events in 11.5%. FD screening found no additional cases among patients with LVNC, besides the previously described case.
ConclusionNo additional FD cases were found among patients with LVNC, which argues against the hypothesis that LVNC is a cardiac manifestation of FD.
Não está esclarecido se a não compactação do ventrículo esquerdo (NCVE) é uma miocardiopatia distinta ou uma manifestação morfológica de várias miocardiopatias. Nós reportamos previamente um caso de NCVE num doente com doença de Fabry (DF), mas permanece por esclarecer se a NCVE é uma manifestação cardíaca de DF, um achado coincidente ou um sobrediagnóstico, o que tem importantes implicações terapêuticas. Este estudo pretende determinar a prevalência de DF em doentes com NCVE.
MétodosEstudo retrospetivo incluindo todos os doentes diagnosticados com NCVE em oito centros hospitalares. O diagnóstico de NCVE foi baseado em pelo menos um dos critérios de ecocardiografia ou ressonância cardíaca. O rastreio de DF foi realizado por teste enzimático e genético.
ResultadosO estudo incluiu 78 doentes diagnosticados com NCVE com base nos critérios de Jenni (84,6%), Stöllberger (46,2%), Chin (21,8%), Petersen (83,8%) e Jacquier (16,2%). A disfunção sistólica do VE estava presente em 48,7%. A insuficiência cardíaca foi encontrada em 60,3%, as disritmias ventriculares em 21,6% e os eventos embólicos em 11,5%. O rastreio de DF não encontrou casos adicionais nos doentes com NCVE, para além do caso previamente descrito.
ConclusãoNão foram encontrados casos adicionais de DF nos doentes com NCVE, o que argumenta contra a hipótese de a NCVE ser uma manifestação cardíaca de DF.
Fabry disease (OMIM 301500) is an X-linked lysosomal storage disorder caused by mutations in the GLA gene, which is located on Xq22.1 and codes for the enzyme alpha-galactosidase A (α-Gal A). Deficiency in the enzyme activity of α-Gal A leads to the lysosomal accumulation of globotriaosylceramide (Gb3) and related glycosphingolipids, causing multiorgan damage.1
Fabry disease is a rare condition, estimated to occur in 1 in 3100 hemizygous males.2 The diagnosis in males is based on biochemical determination of α-Gal A activity. In heterozygous females, α-Gal A activity may be normal, so diagnosis requires molecular analysis of the GLA gene.1 Clinical manifestations may arise early, in childhood or adolescence, and include acroparesthesias, cornea verticillata, angiokeratomas and microalbuminuria or proteinuria. In adulthood, patients may develop sensorineural deafness, renal failure, cerebrovascular events and cardiac manifestations.1
Fabry disease affects the heart, causing left ventricular hypertrophy (LVH) and fibrosis, heart failure, angina, valve thickening or regurgitation, dysrhythmias, cardiac conduction abnormalities and sudden death.1,3 The main cardiac manifestation is hypertrophic cardiomyopathy, which occurs in 43% of male patients (mean age of onset 39±10 years) and 26% of female patients (mean age of onset 50±11 years).4 LVH in Fabry disease is usually non-obstructive and symmetrical3 with prominent papillary muscles5 and, in nearly half of cases, myocardial late gadolinium enhancement is typically observed in the mid-wall of the inferolateral basal segments of the left ventricle.6
Left ventricular noncompaction (LVNC) is morphologically characterized by prominent left ventricular (LV) trabeculations and deep intertrabecular recesses communicating with the LV cavity, located in the apical and lateral regions of the left ventricle, distal to the papillary muscles. The myocardium has a two-layer appearance, composed of a thin compacted epicardial layer and a thick spongy noncompacted endocardial layer.7
LVNC has been found in 0.045% of adult individuals referred for echocardiography8 and 0.029% of children admitted for heart failure due to cardiomyopathy.9 It predominantly affects males8 and individuals of African origin.10 First described on autopsy in 1969 by Feldt et al.11 and on echocardiography by Engberding et al. in 1984,12 it is most commonly diagnosed by transthoracic echocardiography or cardiac magnetic resonance imaging (MRI). Several diagnostic criteria have been proposed using echocardiography or cardiac MRI13–19 (Table 1).
Diagnostic criteria of left ventricular noncompaction.
| Echocardiographic criteria | |
| Jenni et al.13 | (1) Numerous and prominent trabeculations and deep intertrabecular recesses that are localized predominantly in the apical and mid-ventricular segments of the inferior and lateral walls of the left ventricle;(2) Two-layered myocardium, with a thin compacted layer that is adjacent to the epicardium and a much thicker noncompacted layer that is near the endocardium, resulting in a maximum end-systolic ratio of the thickness of noncompacted to compacted layer >2 in short-axis parasternal view;(3) Color Döppler evidence of the perfusion of the intertrabecular recesses directly from the LV cavity;(4) Absence of other cardiac abnormalities. |
| Chin et al.14 | (1) The quotient between the distance from the epicardial surface to the trough of the intertrabecular recess and the distance from the epicardial surface to the peak of the trabeculation decreases from the level of the papillary muscles to the apex and is ≤0.5, at end-diastole, at the level of the LV apex. Measurements are performed in parasternal long-axis view at the level of the mitral valve and papillary muscles and in subxiphoid or apical 4-chamber views at the level of the LV apex. |
| Stöllberger et al.15 | (1) More than three trabeculations protruding from the LV wall, apically to the papillary muscles, are visualized in one imaging plane;(2) The intertrabecular recesses are perfused directly from the LV cavity as shown by color Döppler. |
| Cardiac MRI criteria | |
| Petersen et al.16 | The maximal end-diastolic ratio of the thickness of the noncompacted to the compacted layer, measured in any of the three long-axis views, is >2.3. |
| Jacquier et al.17 | The LV trabeculated mass is over 20% of the total LV mass (papillary muscles are excluded from the trabeculated mass). |
| Stacey et al.18 | The maximal end-systolic ratio of the thickness of the noncompacted to compacted layer in short-axis views is ≥2. |
| Captur et al.19 | Maximal apical fractal dimension >1.3, measured at end-diastole in short-axis views of the apical third of the left ventricle. |
LV: left ventricular; MRI: magnetic resonance imaging.
Patients with LVNC may be asymptomatic or develop complications such as heart failure, thromboembolism or ventricular dysrhythmias and sudden death.20
The etiology and pathogenesis of LVNC are unknown. It has been hypothesized to result from a disturbance of the compaction process of the LV myocardium during early development, but growing evidence suggests that, although the papillary muscles are formed by compaction of pre-existing trabeculations, these trabeculations contribute little, if anything, to the compacted layer of the ventricular walls.21 Moreover, not all cases are congenital and LVNC may be acquired at any stage of life after birth.7
LVNC is characterized by marked genetic heterogeneity. Mutations have been described in genes that code for proteins in the mitochondria (tafazzins), Z line (ZASP/Cypher), cytoskeleton (alpha-dystrobrevin), nuclear lamina (lamin A/C) and sarcomere (beta-myosin heavy chain, alpha-cardiac actin and cardiac troponin T).7 However, the causal relationship between these mutations and LVNC remains to be established.
LVNC has been reported in patients with congenital cardiac abnormalities, such as LV outflow tract abnormalities, Ebstein anomaly, tetralogy of Fallot and double outlet right ventricle; chromosomal defects, such as trisomy 13, Cornelia de Lange, DiGeorge, Turner and 5q minus syndromes; and neuromuscular disorders, such as mitochondrial myopathies, Barth syndrome, zaspopathy, myotonic dystrophy type 1, dystrobrevinopathy, and Emery-Dreifuss muscular dystrophy.7 LVNC has also been reported in patients with storage disorders such as glycogen storage disease types Ib and IV,22,23 Danon disease24 and amyloidosis.25
In 2011, we reported the first case of LVNC in a Fabry disease patient.26 This was a young asymptomatic female who fulfilled the Jenni echocardiographic criteria and the Petersen cardiac MRI criteria for LVNC. She had been diagnosed with Fabry disease following family screening, and the pathogenic mutation of the GLA gene detected was a missense mutation (p.F113L) that is known to be associated with a predominantly cardiac later-onset phenotype. However, thorough diagnostic study revealed none of the known clinical manifestations of Fabry disease, including LVH, late gadolinium enhancement or other cardiac or extracardiac manifestations, nor did she present LV dilation or dysfunction or any clinical complication of LVNC, such as heart failure, dysrhythmias or thromboembolism. Moreover, all of her relatives with Fabry disease underwent echocardiography and cardiac MRI and none presented LVNC. In this report, we pointed out the limitations of the current diagnostic criteria of LVNC and raised the question whether this was an overdiagnosis of LVNC, a coincidental finding of LVNC and Fabry disease, or if indeed the two conditions were clinically related.26,27
More recently, two additional cases of LVNC in Fabry disease patients have been reported,28–31 also in young females. In the second case, the diagnosis of LVNC was based only on cardiac MRI (Petersen criteria), without clear echocardiographic criteria of LVNC due to insufficient quality of the acoustic window. She had classical Fabry disease due to a nonsense GLA gene mutation (p.R220X). As in our case, she also did not present LVH, dilation, dysfunction, late gadolinium enhancement or other cardiac abnormalities, and there was no history of heart failure, dysrhythmias or thromboembolism. There was also no family history of LVNC. She had no clinical manifestations of Fabry disease besides cornea verticillata and angiokeratomas. An endomyocardial biopsy showed deposition of glycosphingolipids in cardiomyocytes and based on this finding the patient was referred for enzyme replacement therapy.28,29
In the third case, the diagnosis of LVNC was based on cardiac MRI (Petersen criteria) and echocardiography (although it was not reported which specific criteria were fulfilled). She was diagnosed with Fabry disease based on low levels of α-Gal A, but the pathogenic GLA gene mutation was never found. The only clinical manifestations of Fabry disease were angiokeratomas and acroparesthesias. As in the two previous cases, there was no LVH, dilation or dysfunction. Cardiac MRI showed subendocardial late gadolinium enhancement, but its location and extension was not reported. Interestingly, she had Wolff-Parkinson-White syndrome, interpreted by the authors as an unrelated finding, and underwent ablation of a para-Hisian accessory pathway, which was followed by disappearance of the short PR interval and delta waves. However, as in the two previous cases, she had no history of heart failure or thromboembolism. Moreover, there were no available data consistent with a family history of LVNC or Fabry disease.30,31
Taken together, it remains essential to clarify whether LVNC could be a cardiac manifestation of Fabry disease, given the implications for therapeutic decisions on whether to start enzyme replacement or chaperone therapy, particularly in young asymptomatic females with no other known clinical manifestations of Fabry disease.
The only published study to date on Fabry disease screening in patients with LVNC included 26 male patients who fulfilled the Stöllberger echocardiographic criteria for LVNC. Fabry disease screening was based on the enzymatic assay of α-Gal A and found no patients with the disease.32
Herein, we report the results of Fabry disease screening in a larger cohort of patients with LVNC.
MethodsPatient populationBetween October 2015 and October 2017, 78 consecutive patients with LVNC were recruited from the cardiology consultation of eight Portuguese hospital centers. The inclusion criteria were adult patients (aged ≥18 years) diagnosed with LVNC based on the presence of any of the current diagnostic criteria of LVNC, by either echocardiography or cardiac MRI, as described in Table 1. All patients fulfilling the inclusion criteria underwent Fabry disease screening.
Fabry disease screeningFabry disease screening was performed at a specialized laboratory (Centogene AG, Rostock, Germany). α-Gal A enzyme activity was measured in dried blood spot samples by fluorometry. Molecular analysis of the GLA gene was performed in males with reduced α-Gal A enzyme activity, i.e., below the normal reference range (<15.3 μmol/l/h), and in all females, independently of α-Gal A activity. The molecular analysis was performed by classical bidirectional Sanger sequencing of the GLA gene and included all exons, exon/intron boundaries and the promoter region.
Ethical issuesThis research project was approved by the ethics committees of all the hospital centers included in this study and all patients provided written informed consent.
ResultsPatients with LVNC were predominantly males (62.8%). Only four patients (5.1%) were identified as being of African origin. Mean age at diagnosis of LVNC was 64.4±14.1 years. A family history of LVNC was recorded in five patients (6.4%). The patients did not present concomitant neuromuscular disorders or any other medical conditions reported to be associated with LVNC.
The diagnosis of LVNC was established by echocardiography in 91.0% of the cases, based on the Jenni criteria in 84.6%, the Stöllberger criteria in 46.2%, the Chin criteria in 21.8% and the three echocardiographic criteria in 20.5% of the cases. Nearly half of the patients presented LV systolic dysfunction (48.7%) and mean LV ejection fraction was 47.2±15.2%. Cardiac MRI was performed in 68 patients (87.2%) and the diagnosis of LVNC was established in 58 patients (85.3%), mainly based on the Petersen criteria (83.8%). The trabeculated LV mass was calculated in only 11 patients, all of them fulfilling the Jacquier criteria of LVNC. The presence of the Stacey or Captur criteria was not assessed in any cardiac MRI. The diagnosis of LVNC was established by both echocardiography and cardiac MRI in 53 of the 68 patients who underwent cardiac MRI (77.9%).
LVNC was an incidental finding in 44.9% of the patients. Heart failure was the most common clinical manifestation at the time of diagnosis (34.6%). In the follow-up (53±33 months), heart failure remained the most frequent clinical complication (60.3%), with New York Heart Association functional class ranging from I to IV (I: 8.5%; II: 59.6%; III: 23.4%; IV: 8.5%). Dysrhythmias were also common (35.9%). Twenty-four-hour Holter monitoring was performed in 74 patients (94.9%) and revealed atrial fibrillation in 10 patients (13.5%) and ventricular tachycardia (VT) in 16 patients (21.6%), although only one case exhibited sustained VT. Thromboembolism occurred in nine patients (11.5%), as stroke in eight and as myocardial infarction in one (Table 2).
Baseline characteristics of patients with left ventricular noncompaction (n=78).
| Gender (male/female) | 49 (62.8%)/29 (37.2%) |
| Race (Caucasian/African) | 74 (94.9%)/4 (5.1%) |
| Age at diagnosis of LVNC, years (mean±SD) | 47±17 |
| Age at Fabry disease screening, years (mean±SD) | 53±17 |
| Family history of LVNC | 5 (6.4%) |
| Family history of Fabry disease | 1 (1.3%) |
| Clinical presentation | |
| Incidental finding | 35 (44.9%) |
| Heart failure | 27 (34.6%) |
| Dysrhythmia | 9 (11.5%) |
| Thromboembolism | 4 (5.1%) |
| Other | 8 (10.3%) |
| Clinical complications in follow-up | |
| Heart failure | 47 (60.3%) |
| Dysrhythmias | 28 (35.9%) |
| Thromboembolism | 9 (11.5%) |
| Diagnosis of LVNC | |
| Echocardiography | 71/78 (91.0%) |
| Cardiac MRI | 58/68 (85.3%) |
| Echocardiographic criteria | |
| Jenni | 66 (84.6%) |
| Stöllberger | 36 (46.2%) |
| Chin | 17 (21.8%) |
| Cardiac MRI criteria | |
| Petersen | 57 (83.8%) |
| Jacquier | 11 (16.2%) |
| Stacey | 0 (0.0%) |
| Captur | 0 (0.0%) |
| Echocardiography | |
| LV ejection fraction, % (mean ± SD) | 47.2±15.2 |
| LV systolic dysfunction | 38 (48.7%) |
| LV diastolic dysfunction | 42 (56.0%) |
| Mean Sa, cm/s (mean ± SD) | 7.7±2.8 |
| Mean Ea, cm/s (mean ± SD) | 9.2±4.1 |
| Mean E/Ea ratio | 8.9±2.7 |
| IVS/PW thickness, mm (mean ± SD) | 9.8±2.9/8.7±1.9 |
| LV mass, g/m2 (mean ± SD) | 121.5±82.3 |
| LA volume, ml/m2 (mean ± SD) | 32.9±13.9 |
| Valve disease | 15 (19.2%) |
| Cardiac MRI | |
| Ratio of thickness of NC:C layer | 3.4±3.9 |
| LV mass, g/m2 (mean ± SD) | 99.5±56.0 |
| LV end-systolic volume, ml/m2 (mean ± SD) | 71.8±50.0 |
| LV end-diastolic volume, ml/m2 (mean ± SD) | 130.1±75.1 |
| LV ejection fraction, % (mean ± SD) | 46.2±16.2 |
| Late gadolinium enhancement | 10 (15.4%) |
| ECG | |
| Sinus rhythm | 70 (89.7%) |
| Atrial fibrillation | 6 (7.7%) |
| Atrial flutter | 2 (2.6%) |
| AV block | 4 (5.1%) |
| LBBB | 10 (12.8%) |
| RBBB | 3 (3.8%) |
| Bifascicular block | 2 (2.6%) |
| 24-hour Holter | |
| Sinus rhythm | 67 (90.5%) |
| Atrial fibrillation | 10 (13.5%) |
| Atrial flutter | 1 (1.4%) |
| AV block | 12 (16.2%) |
| LBBB | 11 (14.9%) |
| RBBB | 3 (4.1%) |
| Bifascicular block | 2 (2.7%) |
| Supraventricular tachycardia | 13 (17.6%) |
| VT | 16 (21.6%) |
| Non-sustained VT | 15 (20.3%) |
| Sustained VT | 1 (1.4%) |
| Clinical manifestations of Fabry disease | |
| Renal failure | 7 (9.0%) |
| Microalbuminuria | 1 (1.3%) |
| Proteinuria | 0 (0.0%) |
| Stroke | 8 (10.3%) |
| Acroparesthesias | 1 (1.3%) |
| Deafness | 2 (2.6%) |
AV: atrioventricular; C: compacted; ECG: electrocardiogram; IVS: interventricular septum; LA: left atrial; LBBB: left bundle branch block; LV: left ventricular; LVNC: left ventricular noncompaction; MRI: magnetic resonance imaging; NC: non-compacted; PW: posterior wall; RBBB: right bundle branch block; VT: ventricular tachycardia.
Twenty-eight patients (35.9%) had undergone genetic study that included a wide range of genes (MYH7: 32.1%; MYBPC3: 30.8%; TNNT2: 28.2%; TNNI3: 19.2%; ACTC1: 28.2%; TPM1: 28.2%; MYL2: 11.5%; MYL3: 11.5%; DTNA: 7.7%; DMD: 10.3%; LDB3: 28.2%; TAZ: 26.9%; LMNA: 24.4%; CSRP3: 24.4%; TCAP: 20.5%; SGCD: 21.8%; PLN: 23.1%; TTN: 2.6%; SCN5A: 1.3%). A pathogenic mutation was found in the MYH7 gene in one patient (c.1003G>C) (unpublished data). Eight genetic variants of uncertain significance were found in six cases: four in MYBPC3 (c.1505G>A; c.2762A>T; c.1624G>C; c.3676C>T), the last two in the same patient; two in TTN (c.16425T>G; c.60259C>G); one in SCN5A (c.393-5C>A); and one in SGCD (c.772-774delAAG) in the same patient carrying two genetic variants of the MYBPC3 gene.
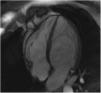
The screening of 78 patients with LVNC revealed no additional cases of Fabry disease besides the one already identified.26 This Fabry patient was never under enzyme replacement or chaperone therapy. During the eight-year follow-up, this patient has remained asymptomatic and without known clinical manifestation of Fabry disease or complication of LVNC. Moreover, there were no significant changes regarding the location or extension of LVNC or LV chamber size, mass or function during follow-up (Figure 1).
Discussion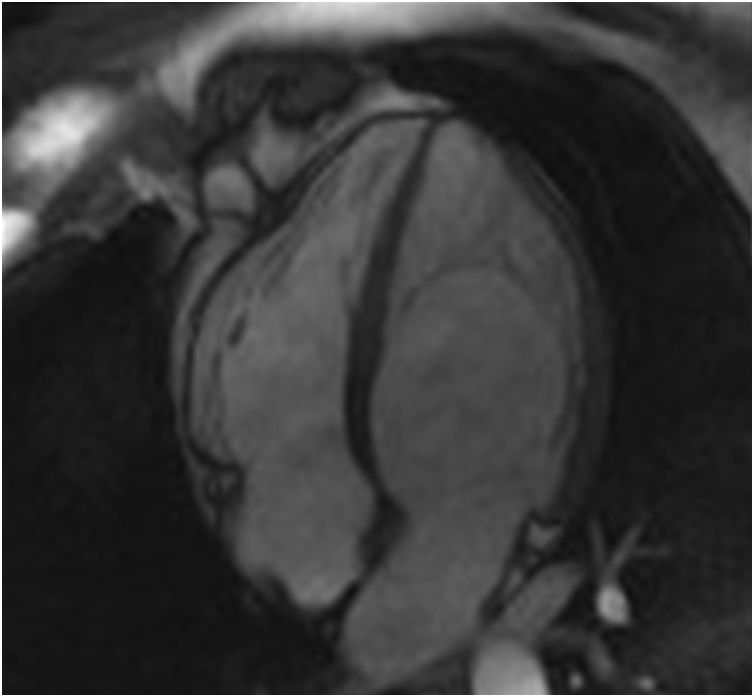
This multicenter study of Fabry disease screening in 78 consecutive patients with LVNC revealed no additional Fabry patients besides the single case that had been previously reported by our center, which argues against the hypothesis that LVNC is a clinical manifestation of Fabry disease.
The most likely explanation for the finding of LVNC in this Fabry patient is an overdiagnosis of LVNC. Studies have increasingly exposed the lack of specificity of the current diagnostic criteria for LVNC, potentially leading to overdiagnosis.10,33,34 Moreover, there is growing evidence that LVNC has no independent prognostic impact compared to conventional risk factors, such as LV dilation, dysfunction or fibrosis, and that patients with LVNC without LV dilation, dysfunction or fibrosis have an excellent prognosis.34–36
In a study of 199 patients referred to a heart failure clinic and 60 healthy controls, Kohli et al. found that 23.6% of the patients and 8.3% of the controls fulfilled one or more echocardiographic criteria for LVNC. Among the heart failure patients with LVNC criteria, 29.8% fulfilled all three echocardiographic criteria and 36.2% fulfilled only a single criterion, with the frequency of each criterion ranging from 12.1% to 18.6%.10 In a large population-based cohort of 1000 subjects from the Multi-Ethnic Study of Atherosclerosis (MESA) registry who underwent cardiac MRI, the Petersen criteria for LVNC were fulfilled in 43% of the subjects without cardiac disease.33 Another study based on the MESA registry including 2742 subjects undergoing cardiac MRI showed that a greater extent of LV trabeculations in asymptomatic individuals appears to be benign and is not associated with deterioration in LV volumes or function over an almost 10-year period.36 In a study including 700 patients referred for cardiac MRI, Ivanov et al. found an LVNC prevalence of 39%, 23%, 25% and 3%, according to the Petersen, Stacey, Jacquier and Captur criteria, respectively. In this study, during a median follow-up of seven years, there were no statistically significant differences in the assessed outcomes (death, ischemic stroke, ventricular tachycardia/fibrillation, and heart failure hospitalization) between patients with and without LVNC, irrespective of the criteria applied.34 Finally, in a study including 113 patients with LVNC according to the Jenni and Petersen criteria, Andreini et al. found that the degree of hypertrabeculation had no prognostic impact over and above LV dilation, dysfunction or late gadolinium enhancement. Indeed, patients without LV dilation, dysfunction or fibrosis had an excellent prognosis at four-year follow-up.35
In light of this evidence, the possibility of an overdiagnosis of LVNC, with no associated prognostic impact, must be considered in these three cases of Fabry disease published in the literature. In fact, all three case reports of LVNC occurred in young females with no family history of LVNC in Fabry or non-Fabry relatives, no LV hypertrophy, dilation or dysfunction, no late gadolinium enhancement suggestive of Fabry disease involvement, no or few clinical manifestations of Fabry disease, and no clinical complications of LVNC.26–31
Appropriate follow-up would also be required to correctly interpret the significance of LVNC in these Fabry patients. In our case, the patient was never under enzyme replacement or chaperone therapy, because at baseline she did not present any known manifestation of Fabry disease that would constitute a clinical indication to start specific therapy according to the guidelines. During the eight-year follow-up of this patient, no significant change has been documented in the location or extension of LVNC or in LV chamber size, mass or function. Moreover, this patient has remained asymptomatic and with no known clinical manifestation of Fabry disease or complication of LVNC, which supports the hypothesis of an overdiagnosis of LVNC in this Fabry patient.
Another possibility is that LVNC and Fabry disease in these patients are coincidental and unrelated conditions. Although the simultaneous finding of two unrelated rare conditions may seem unlikely, it is now known that LVNC has a much higher prevalence10,33,34 than the previous estimate of 0.045%8 and the currently estimated Fabry disease prevalence of 1 in 31002 is much higher than the previous estimate of 1 in 117 000.37
The finding of LVNC in a wide variety of congenital cardiac abnormalities, chromosomal defects, neuromuscular disorders7 and storage disorders,22–24 as well as in elite athletes38 and pregnant women,39 has raised the question of whether LVNC is indeed a distinct cardiomyopathy or simply a common epiphenomenon in cardiac overload conditions. In our case, this young woman was not an athlete, but she had been pregnant once, although we have no record of echocardiograms before, during or just after pregnancy. A history of pregnancy or vigorous exercise was not reported in the other two cases.
Finally, there is the possibility that LVNC could be a cardiac manifestation of Fabry disease. However, there is no evidence that Fabry disease causes LVNC by either compensatory or genetic mechanisms. Indeed, although they had Fabry disease, none of these patients had concomitant LVH, dilation or dysfunction that could have led to cardiac overload and hence to LVNC. Furthermore, there is no evidence that the GLA gene mutation, the mutated α-Gal A, or the intramyocardial deposition of glycosphingolipids due to Fabry disease are the cause of or are in any way related to the development of LVNC, and any assumption on this point remains speculative. Lysosomal accumulation of Gb3 begins in utero and has been demonstrated as early as the 17th week of gestation in various fetal cell types, such as cardiomyocytes,40 kidney podocytes,40–42 hepatocytes,41,43 corneal epithelial cells44 and myenteric plexus cells.42 Although Gb3 storage begins in utero, clinical manifestations only develop several years later. Therefore, Gb3 deposits are expected findings in biopsies, including endomyocardial biopsies, of all Fabry patients independently of the presence of clinical manifestations, and such deposition should not be interpreted as evidence that LVNC is due to Fabry disease.
The only study screening for Fabry disease in patients with LVNC did not reveal any positive case. However, this was a small cohort of 26 male patients and the only criteria for LVNC diagnosis were the Stöllberger echocardiographic criteria.32 Our study, on the other hand, included a larger cohort of patients and the inclusion criteria allowed (LVNC) diagnosis based on one or more criteria, by echocardiography or cardiac MRI. Our cohort of patients appears to be representative of LVNC and its wide clinical spectrum. Although a significant proportion of cases were incidental findings, there was good agreement between echocardiography and cardiac MRI regarding the diagnosis of LVNC, nearly half of the patients presented LV systolic dysfunction, and in the follow-up most patients developed complications. Even so, no Fabry disease patient was diagnosed besides the single case already reported in the literature. Nevertheless, our study is also limited by its small sample size, which may have prevented the detection of Fabry disease if LVNC was a rare manifestation of this disease. However, the common finding of LVNC in other patient populations, such as heart failure, also argues against the hypothesis of LVNC being a rare clinical manifestation of Fabry disease. The fact that our patient remained stable and did not develop any complications during an eight-year follow-up without specific therapy for Fabry disease also argues against the hypothesis that LVNC is a clinical manifestation or a risk marker for the development of other cardiac manifestations of Fabry disease.
Based on recent evidence exposing the low specificity of the current diagnostic criteria and the increasing rate of overdiagnosis of LVNC,10,33,34 it is plausible that more cases of LVNC will be diagnosed among Fabry patients, raising concerns about its therapeutic implications. This is particularly important in Fabry females, who, in part due to the inactivation pattern of the X chromosome, present a highly variable prognosis, some of them exhibiting a benign disease course and a lifelong prognosis without significant clinical manifestations in the absence of specific therapy.45
ConclusionIn summary, our study found no evidence to support the hypothesis that LVNC is a clinical manifestation of Fabry disease. Taken together with the available evidence, our results do not support the use of the current diagnostic criteria of LVNC for the decision to start specific therapy for Fabry disease, either enzyme replacement or chaperone therapy, in young asymptomatic females presenting LVNC criteria in the absence of other cardiac abnormalities or known manifestations of Fabry disease that would constitute a clinical indication to start therapy according to the current guidelines.
DisclosuresOlga Azevedo, MD, and Gabriel Miltenberger-Miltenyi, PhD, have received educational and/or research grants from Shire Human Genetic Therapies, Inc., Genzyme Corporation and Amicus Nuno Marques, MD, Hugo Antunes, MD, and Liliana Reis, MD, have received educational grants from Shire Human Genetic Therapies, Inc. and Genzyme Corporation. Nuno Craveiro, MD, Rui Azevedo Guerreiro, MD, and Rui Pontes dos Santos, MD, have received educational grants from Shire Human Genetic Therapies, Inc. The other authors have no disclosures.
FundingThis research was supported by a research grant from Shire Human Genetic Therapies, Inc.
Shire Human Genetic Therapies, Inc. had no influence on the design and conduct of the study, in the collection, analysis, and interpretation of the data, or in the preparation, review, or approval of the manuscript.
Conflicts of interestThe authors have no conflicts of interest to declare.
