






The myocardial response to acute stretch consists of a two-phase increase in contractility: an acute increase by the Frank-Starling mechanism and a gradual and time-dependent increase in force generated known as the slow force response (SFR). The SFR is actively modulated by different signaling pathways, but the role of protein kinase G (PKG) signaling is unknown. In this study we aim to characterize the role of the PKG signaling pathway in the SFR under normal and ischemic conditions.
MethodsRabbit papillary muscles were stretched from 92 to 100% of maximum length (Lmax) under basal conditions, in the absence (1) or presence of: a PKG agonist (2) and a PKG inhibitor (3); under ischemic conditions in the absence (4) or presence of: a PKG agonist (5); a nitric oxide (NO) donor (6) and a phosphodiesterase 5 (PDE5) inhibitor (7).
ResultsUnder normoxia, the SFR was significantly attenuated by inhibition of PKG and remained unaltered with PKG activation. Ischemia induced a progressive decrease in myocardial contractility after stretch. Neither the PKG agonist nor the NO donor altered the myocardial response to stretch under ischemic conditions. However, the use of a PDE5 inhibitor in ischemia partially reversed the progressive deterioration in contractility.
ConclusionsPKG activity is essential for the SFR. During ischemia, a progressive decline in the force is observed in response to acute myocardial stretch. This dysfunctional response can be partially reversed by the use of PDE5 inhibitors.
A resposta ao estiramento agudo do miocárdio consiste num aumento bifásico da contractilidade: um aumento agudo pelo mecanismo de Frank-Starling e um aumento gradual denominado resposta inotrópica tardia (SFR). A SFR é modulável por diferentes vias de sinalização, no entanto, o papel da via da Proteína Cínase G (PKG) permanece desconhecido. Assim, no presente estudo, pretendemos caracterizar o papel da via de sinalização da PKG na SFR em condições normais e em condições isquémicas.
MétodosMúsculos papilares de coelho foram estirados de 92 para 100% de Lmax em condições basais, na ausência (1) ou na presença de: um agonista da PKG (2) e um inibidor da PKG (3); em condições isquémicas, na ausência (4) ou na presença de: um agonista da PKG (5), um dador de óxido nítrico (NO) (6) e um inibidor da fosfodiesterase 5 (PDE5) (7).
ResultadosEm condições basais, a SFR foi significativamente atenuada pela inibição da PKG, não tendo sido alterada pela ativação da PKG. A isquemia induziu uma diminuição progressiva da contratilidade após o estiramento agudo. Esta resposta não foi alterada pela adição de agonista da PKG nem pelo uso de um dador de NO. No entanto, o uso de um inibidor da PDE5 durante a isquemia foi capaz de reverter parcialmente a deterioração progressiva da contractilidade.
ConclusõesA atividade da PKG é essencial para a SFR. Durante a isquemia observa-se uma diminuição progressiva da contratilidade em resposta ao estiramento agudo. Esta resposta disfuncional pode ser revertida pelo uso de inibidores da PDE5.
active tension
cyclic guanosine monophosphate
end-diastolic volume
Frank-Starling mechanism
nitric oxide
phosphodiesterase
protein kinase G
slow force response
troponin I
The heart is able to adapt continuously to ever-changing hemodynamic conditions. An acute hemodynamic overload, such as the beginning of aerobic exercise, increases venous return, eliciting myocardial stretch and an increase in end-diastolic volume (EDV) and pressure. The systolic arm of the response to myocardial stretch was first described over a century ago by Ernest Starling and Otto Frank,1–5 who discovered that increased EDV promotes an immediate increase in contractility and stroke volume, the Frank-Starling mechanism (FSM). After the initial response, there is a progressive and time-dependent increase in contractility, first described in 1912 by von Anrep6 and later explored by Parmley and Chuck in 1973.7 In vivo, this mechanism allows the return of EDV to its initial value and is known as the von Anrep effect. Its in-vitro counterpart is known as the slow force response (SFR).
The SFR is a consequence of an increase in magnitude of calcium transients, due to activation of the Na+/H+ antiporter. This mechanism favors the reverse operating mode of the Na+/Ca2+ antiporter, leading to an increase in intracellular calcium.8–10 This phase of the myocardial response to stretch is known to be dependent on various signaling pathways and acutely modulated by various neurohumoral mediators.8,9,11 Despite the importance of the PKG signaling pathway in cardiovascular homeostasis, its role in the SFR still mostly unexplored. Pharmacological modulation of this pathway, mostly by drugs that activate NO pathways (nitrates), is extremely common in ischemic heart disease and myocardial infarction. Recently, there has been considerable interest in the beneficial effects of phosphodiesterase 5 (PDE5) inhibitors in the treatment of acute myocardial ischemia.12,13 Myocardial ischemia promotes contractile dysfunction and acute hemodynamic overload, thereby leading to myocardial stretch. Therefore, discerning the influence of the PKG pathway on the contractile response to stretch under ischemic conditions and, particularly, its pharmacological modulation (by NO donors and PDE5 inhibitors) is central to more accurate treatment decisions.
MethodsModulation of the PKG signaling pathway during acute myocardial stretchThe effects of acute muscle stretch from 92 to 100% of maximum length (Lmax) on contractile function of rabbit papillary muscles were assessed under basal conditions, in the absence (control group, n=8) or the presence of a cell-permeable PKG activator (8-bromo-cGMP, 10−6 M; PKGa group, n=7) and a PKG inhibitor (Rp-8-Br-PET-cGMPS, 10−6 M; PKGi group, n=7). These substances were dissolved in Krebs-Ringer solution and myocardial stretch was elicited 20 min later.
Modulation of the PKG signaling pathway during acute myocardial stretch under ischemic conditionsIn a second set of protocols, the contractile response to stretch was studied under ischemic conditions. After stabilization, the bathing solution was replaced by another without glucose and without O2 supply. After 5 min of ischemia, muscles were stretched from 92 to 100% of Lmax (Isch group, n=8). Furthermore, to explore the influence of modulation of the PKG pathway on the systolic response to stretch during ischemia, the latter protocol was also performed in the presence of a PKG activator (8-bromo-cGMP, 10−6 M); Isch-PKGa group, n=7; (H) an NO donor (S-nitroso-N-acetyl-penicillamine (SNAP), 10−5 M; Isch-NO group, n=9) and (I) a PDE5 inhibitor (sildenafil, 10−6 M; Isch-PDE5i group, n=7). These drugs interfere with the NO/PKG pathway at different targets and allow better characterization of SFR modulation. Ischemia was maintained for 15 min. To confirm myocardial viability after the ischemic period, the muscles were reperfused with a glucose-containing Krebs-Ringer solution and O2 supply.
A schematic representation of the protocols is shown in Figure 1.
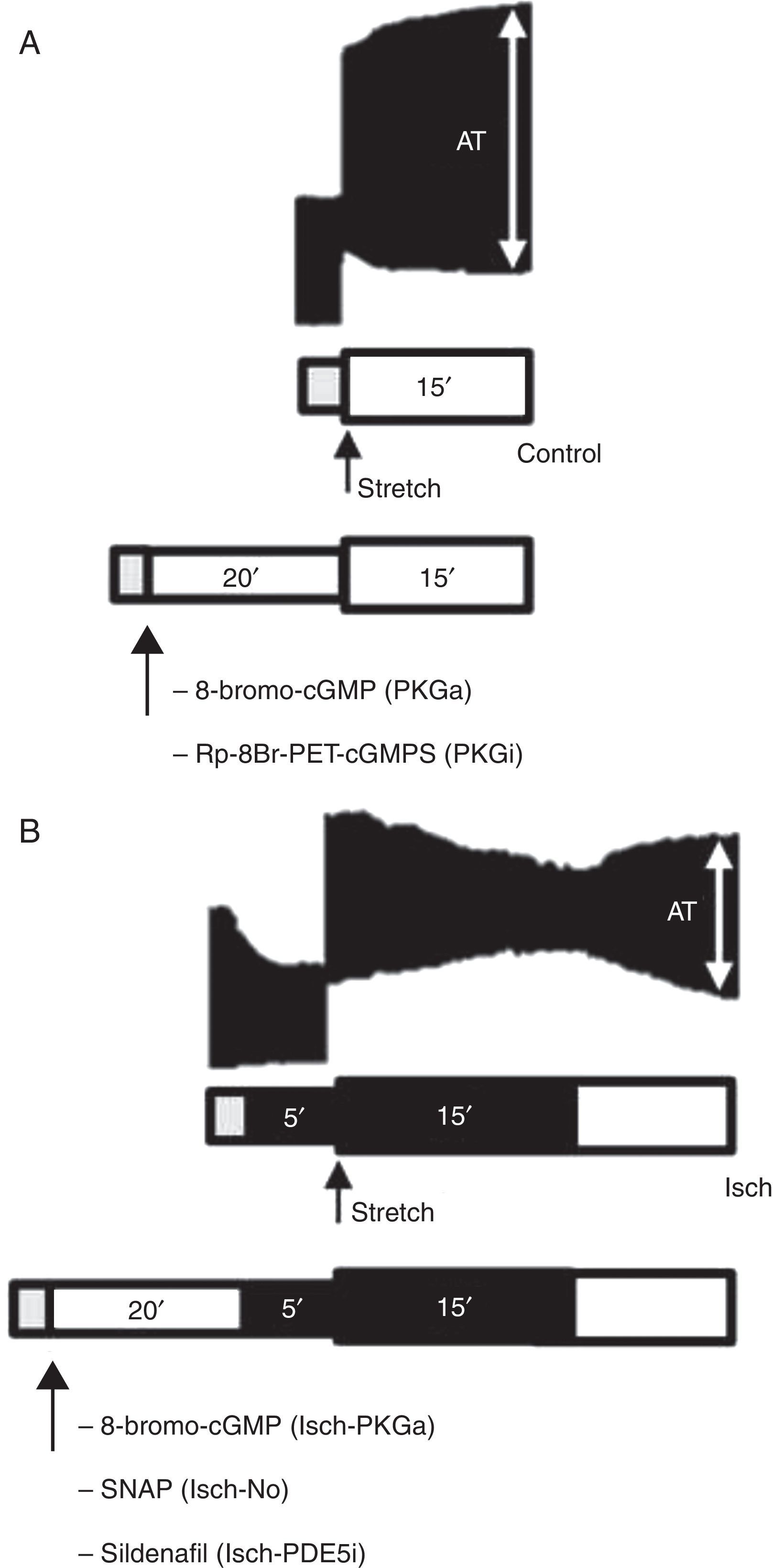
Representative bands of the contractile response to acute stretch (upper images) in the control group (A) and Isch group (B) and schematic illustration of experimental protocols (bottom diagrams). Gray bar: stabilization period; white bar: presence of O2 and glucose; dark bar: absence of O2 and glucose (ischemia). AT: active tension.
The doses were selected according to the literature data and our preliminary studies. All chemicals were obtained from Sigma-Aldrich (St. Louis, MO, USA).
Statistical analysisValues are means±standard error. Within the same group, the effects of acute muscle stretch on the various experimental parameters at specific time points were analyzed with a paired Student's t test. The time-dependent effects of muscle stretch within the same group were analyzed by repeated measures one-way ANOVA, while repeated measures two-way ANOVA was used to compare the time-dependent effects of muscle stretch between several groups. When significant differences were detected with any of the ANOVA tests, the Holm-Sidak method was used to perform multiple comparisons. A value of p<0.05 was accepted as significant.
ResultsBasal pharmacological effectsThe muscles stabilized in the presence of the drug for 20 min, during which no significant variations in active tension were observed.
Contractile response to acute stretch in normal myocardiumIn the control group, active tension (AT) at Lmax, following stretch, was 23.4±3.8 mN/mm2. In the following 15 min there was a progressive increase in AT to 29.1±4.3 mN/mm2 (p<0.001), corresponding to an SFR of 27.8±3.9%. A representative band of the contractile response of a muscle in the control group is shown in Figure 1A.
PKG-mediated effectsIn the PKGa group no differences were found in SFR after 15 min (20.9±8.13% vs. 27.8±3.9%, p=0.324) (Figure 2). On the other hand, myocardial stretch in the presence of the PKG inhibitor was associated with a significant attenuation of SFR (SFR at 15 min: 11.5±5.9% vs. 27.8±3.9% in controls, p=0.034) (Figure 2).
Adaptation of contractile function to myocardial stretch under ischemic conditions SFR by
SFR by In the first 5 min of ischemia a significant decrease in myocardial contractility of 51.0±4.0% was seen (AT decreased from 20.1±3.1 mN/mm2 to 10.2±2.2 mN/mm2, p<0.001). After stretch, muscles did not display SFR; instead, they exhibited a significant and progressive decrease in contractility (AT decreased from 12.4±2.3 mN/mm2 to 5.7±1.9 mN/mm2, p<0.001) (Figure 3). A representative band of a muscle of the Isch group is shown in the Figure 1B.
NO-mediated effects under ischemic conditions SFR by ischemia. In the Isch group, muscles did not display
SFR by ischemia. In the Isch group, muscles did not display The use of an NO donor did not significantly influence the myocardial response to acute stretch under ischemic conditions (AT variation after stretch: −27.7±15.2% in Isch-NO vs. −52.4±7.9% in Isch, p=0.229) (Figure 4).
PKG-mediated effects under ischemic conditions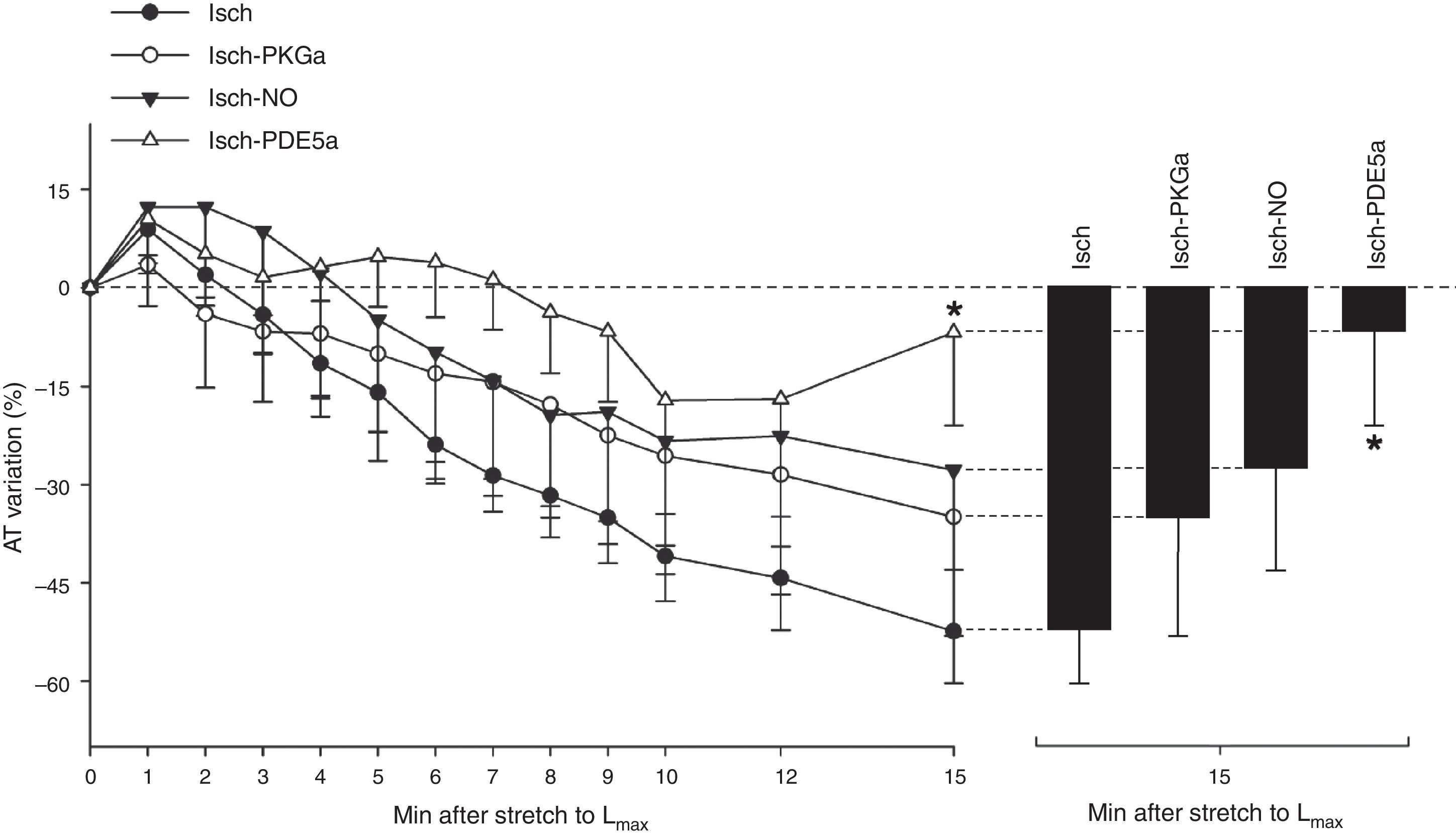
The decrease in contractility over the 15 min after stretch was not significantly different in the presence of the PKG activator compared to the Isch group (−34.9±18.2% vs. −52.4±7.9% in Isch, p=0.373) (Figure 4).
PDE5 inhibition under ischemic conditionsIn contrast to Isch-PKGa, PDE5 inhibition significantly prevented the decrease in contractility after stretch (−8.6±17.6% vs. −52.4±7.9% in Isch, p=0.018) (Figure 4).
DiscussionThe effect of ischemia on myocardial response to stretchAcute myocardial ischemia induces immediate contractile dysfunction, resulting in sudden hemodynamic overload. Hence, it is interesting to characterize the myocardial response to stretch under ischemic conditions. Despite its importance, there is limited knowledge about the effect of ischemia on the myocardial adaptation to stretch. We reported a reversed SFR in ischemic myocardium. This observation may be related to the profound changes in calcium sensitivity observed during ischemia.14,15 Other mechanisms contributing to this dysfunction may include the limited availability of adenosine triphosphate under ischemic conditions, which is known to cause an immediate increase in contractility16 and to promote the development of myocardial rigor due to impairment of sarcomere cross-bridge cycling.17,18
PKG signaling pathways in the contractile response to stretchWe describe some interesting results regarding the role of PKG in the myocardial response to stretch. Our work demonstrates that inhibition of PKG leads to reduced SFR, but activation does not affect this response. These results suggest a permissive role of the PKG signaling pathway in the normal SFR. These results can be explained by a dynamic interaction between PKG and PDE (Figure 5). Under normal conditions, the basal concentration of cGMP in the human myocardium inhibits PDE3, thus preventing the hydrolysis of cAMP,19,20 the presence of which contributes to the normal SFR.21 We therefore hypothesize that removal of the basal PKG pathway increases the action of PDE3, reducing cAMP levels and thereby the SFR. Another possible mechanism linking PKG activity to the SFR is the troponin I (TnI) phosphorylation level. PKG is known to phosphorylate TnI at serine 23/24, increasing the cross-bridge cycling rate,22 which probably contributes to a more efficient SFR.23
 PKG signaling pathway in the normal
PKG signaling pathway in the normal One of the most intriguing results of our work is the observation that sildenafil, but neither 8-bromo-cGMP nor an NO donor significantly prevented the gradual decrease in contractility after acute stretch in ischemia. As the best-characterized action of sildenafil is PDE5 inhibition, preventing cGMP degradation, its effects would be expected to be similar to those of a cGMP analog. Interestingly, data published by Elrod et al.24 suggest a cardioprotective role for sildenafil, independent of its action on the NO/cGMP pathway. It is possible that the contractile preservation in ischemia induced by sildenafil is dependent on a still obscure effect of this molecule, independent of its action on PDE5.
Our findings in the rabbit myocardium should be extrapolated to other species with caution. The known intracellular mechanisms of the SFR depend on the animal species under consideration.10,25,26 However, the increase in calcium transients due to activation of the Na+/H+ exchanger and reverse operating mode of the Na+/Ca2+ exchanger seems to be angiotensin-II independent in both rabbit and human myocardium, depending on acute mechanical stretch alone.25,26 In this way, it is probably easier to extrapolate the findings of this work to the human myocardium than to the other commonly used animal models such as cat, rat, and mouse.
Conclusions and potential therapeutic implicationsIn this work, we evaluated the contractile adaptation to acute stretch in the presence of modulators of PKG signaling pathways, crucial therapeutic weapons in ischemic heart disease and myocardial infarction. From a clinical perspective, we showed that the well-known protective effect of nitrates during an ischemic insult is not associated with further contractile deterioration in response to the hemodynamic overload that typically occurs during cardiac ischemia. We also described an interesting and previously unknown protective effect of sildenafil on the contractile function of the heart following stretch in ischemic conditions. We can thus conjecture that patients chronically using this PDE5 inhibitor may exhibit a better cardiac performance during an ischemic heart event.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that the procedures followed were in accordance with the regulations of the relevant clinical research ethics committee and with those of the Code of Ethics of the World Medical Association (Declaration of Helsinki).
Confidentiality of dataThe authors declare that no patient data appear in this article.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflicts of interestThe authors have no conflicts of interest to declare.
We wish to thank Marta Oliveira for her technical assistance.
These authors equally contributed to this article.
Supported by the Portuguese Foundation for Science and Technology Grants PEst-C/SAU/UI0051/2011 and EXCL/BIM-MEC/0055/2012 through the Cardiovascular R&D Unit and by European Commission Grant FP7-Health-2010; MEDIA-261409.
