








Cardiomyocyte hypertrophy is an important feature of hypertension. However, its molecular underpinnings, especially the signaling cascades, remain unclear. Here we hypothesized that a protein kinase D (PKD)-dependent extracellular signal-regulated kinase 5 (ERK5) pathway was able to regulate downstream myocyte enhancer factor 2D (MEF2D), affecting prohypertrophic responses to angiotensin II (Ang II).
MethodsNeonatal rat cardiomyocytes from 2- to 3-day-old Sprague-Dawley rats were prepared and Western blot, real-time quantitative PCR and immunofluorescence staining were used to assess the activation and translocation of pathway signaling molecules. Atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) expression and [3H]-leucine (Leu) incorporation were measured to determine cell hypertrophy.
ResultsElevated levels of phosphorylated PKD (p-PKD) and ERK5 (p-ERK5) were observed in cardiomyocytes stimulated with Ang II, while silencing protein kinase C epsilon (PKCɛ) resulted in significantly lower levels of p-PKD. Furthermore, Ang II-induced ERK5 activated translocation was mediated by the PKD pathway. Consequently, inhibiting PKCɛ, PKD and ERK5 by siRNA significantly attenuated Ang II-induced MEF2D activation, ANP and BNP mRNA expression, and [3H]-Leu incorporation.
ConclusionsOur studies are the first to show that the PKCɛ/PKD/ERK5/MEF2D pathway plays an important role in the cardiomyocyte hypertrophy response to Ang II.
A hipertrofia dos cardiomiócitos é uma característica importante da hipertensão arterial. No entanto, os mecanismos moleculares da hipertrofia, especialmente as cascatas sinalizadoras, são ainda pouco claros. Neste contexto colocamos a hipótese de que a proteína quinase D (PQD) – dependente da via da quinase 5 (ERK5) determinada pelo sinal extracelular –possa regular a jusante o fator intensificador do miócito -2D (FIM2D), afetando as respostas pro-hipertróficas à Ang II.
MétodosForam preparados os cardiomiócitos de ratos neonatais de dois a três anos, Sprague-Dawley, tendo sido utilizados os testes de Western blot, a PCR quantitaiva Real-Time (PCRq-RTRT) e a coloração por imunofluorescência para avaliar a ativação e a translocação das moléculas das vias de sinalização. O fator natriurético auricular (FNA), o peptídeo natriurético auricular tipo B (BNP) e a incorporação [3H]-Leu foram igualmente medidos para determinar a hipertrofia celular.
ResultadosNos cardiomiócitos estimulados com Ang II foram observados níveis elevados de PCD fosforilada (PCD-f) e ERK5 (ERK5-p), tendo a proteínaquinase C epsilon silenciada apresentado níveis significativamente inferiores de PQD-f. Além disso, a translocação ativada de ERK5 induzida pela Ang II foi mediada pela via PCD. Consequentemente, a inibição de PCDɛ, de PCD e de ERK5 através de siRNA atenuou significativamente a ativação de FIM2D induzida pela Ang II, o FNA e a expressão de mRNA BNP, bem como a incorporação de Leucina-[3H].
ConclusõesO nosso estudo demonstra pela primeira vez que as vias PCCɛ/PCD/ERK5/FIM 2D desempenham um papel importante na resposta hipertrófica dos cardiomiócitos a Ang II.
The onset of heart failure is typically preceded by cardiac hypertrophy, a response of the heart to increased workload, a cardiac insult such as a heart attack, or a genetic mutation. Hypertrophy initially develops as an adaptive response to physiological and pathological stimuli, but pathological hypertrophy generally progresses to heart failure. Cardiac hypertrophy is usually characterized by cardiomyocyte hypertrophy and thickening of ventricular walls.1,2 Although treating various pathways and targets has been reported to be effective, pathological cardiac hypertrophy inevitably leads to the unfavorable outcomes of heart failure.3,4 It is therefore important to find novel therapeutic targets for hypertrophy.
Protein kinase D (PKD) has unique structural, enzymatic and regulatory properties that are different from those of the PKC family members. In unstimulated cells, PKD is in a state of low catalytic kinase activity. In response to cellular stimuli, it is converted into a form with high catalytic activity via phosphorylation at Ser744/748 in the activation loop of the PKD catalytic domain as well as the autophosphorylation of Ser916. PKD has been best characterized for its role in regulating myocardial contraction, cardiac hypertrophy and remodeling.5,6 PKD is a typical histone deacetylase 5 (HDAC5) kinase and thus more relevant to HDAC5-myocyte enhancer factor 2 (MEF2) hypertrophic signaling.7 Mice with PKD ablation show substantial resistance to cardiac hypertrophy.8
Recent studies have implicated extracellular signal-regulated protein kinase 5 (ERK5) as a potential signal transducer for cardiac hypertrophy.9 Activated ERK5 transfers from cytoplasm to nucleus, where it exhibits transcriptional regulatory activity by phosphorylating MEF2.10 Notably, the transcription factor MEF2 promotes cardiac hypertrophy in transgenic mice, while targeted deletion of this kinase attenuates the hypertrophic response mediated by MEF2 activation in vivo.11,12 Our previous studies have indicated that protein kinase C epsilon (PKCɛ)-dependent ERK5 activation and nuclear translocation is involved in cardiomyocyte hypertrophy induced by angiotensin II (Ang II).13
The aim of this study was to determine the potential role of PKD in Ang II-mediated cardiomyocyte hypertrophy. The results presented here demonstrate that Ang II rapidly induces activation of PKD via PKCɛ, which subsequently leads to ERK5-activated translocation, myocyte enhancer factor 2D (MEF2D) transcriptional activation, and cardiomyocyte hypertrophy. Based on our findings, we suggest that the PKCɛ/PKD/ERK5/MEF2D pathway is implicated in Ang II-induced cardiomyocyte hypertrophy.
MethodsMaterialsAll basic laboratory reagents and Ang II were obtained from Sigma-Aldrich (St. Louis, MO). Nuclear and cytoplasmic protein extraction reagents were purchased from Thermo Fisher Scientific (Waltham, MA). Antibodies against β-actin and lamin B1 were purchased from Proteintech Group Inc. (Rosemont, IL). Antibodies against PKD, MEF2D, phospho-PKD744/748, phospho-PKD916, and phospho-MEF2D were obtained from Cell Signaling Technology (Cambridge, MA); antibodies against PKCɛ, ERK5, and phospho-ERK5 from Cayman Chemicals (Ann Arbor, MI), SDS-polyacrylamide gels from Pierce (Rockford, IL), and PVDF and proteingel apparatus from Bio-Rad (Hercules, CA). Minimal essential medium (MEM), fetal bovine serum, phosphate buffered saline (PBS), penicillin/streptomycin, non-essential amino acids, and l-glutamine were obtained from Sigma-Aldrich (St. Louis, MO).
Cell culturesAnimals used in these experiments were handled in accordance with the Guiding Principles in the Care and Use of Animals, approved by the experimental animal ethics committee of Shandong Provincial Hospital Affiliated to Shandong First Medical University. Neonatal rat cardiomyocytes from 2- to 3-day-old Sprague-Dawley rats were prepared as previously described.14 Briefly, cells were obtained by trypsinization after gentle mechanical disruption and grown in MEM (5 ml penicillin/streptomycin, 5 ml non-essential amino acids, 5 ml l-glutamine and 10% FBS in 500 ml of MEM) at 37°C and 5% CO2. Cardiomyocytes were cultured as monolayers at 5×104 cells/cm2 and incubated with Ang II at different times and various dosages. Prior to stimulation, all cells were placed in serum-reduced medium (0.1% FBS) for 24 h to maintain quiescence.
Nuclear and cytoplasmic fractionationNuclear and cytoplasmic protein extraction reagents were used according to the manufacturer's instructions. Cells were washed with cold PBS, suspended in ice-cold buffer F (210 mM mannitol, 70 mM sucrose, 5 mM Tris, pH 7.5, 1 mM EDTA, supplemented with protease inhibitors), left in the buffer for 15 min on ice and then Dounce homogenized (15 strokes). The nuclei were separated by centrifugation (500 g, 5 min, 4°C). The supernatant containing the cytosolic fraction was boiled in sample buffer. The pellet containing the nuclei was washed with PBS and then resuspended in RIPA buffer for 5 min on ice, centrifuged again and the supernatant (nuclear fraction) was boiled in sample buffer. The same volumes of nuclear and cytosolic fractions were analyzed.
Western blot analysisCardiomyocytes were lysed in ice-cold lysis buffer (150 mM NaCl, 5 mM MgCl2, 1 mM phenylmethylsulfonyl fluoride, 1 mM dithiothreitol, 1 mM sodium orthovanadate, 1 mM sodium phosphate, 1% Triton X-100, 0.5% SDS, 10 μg/ml aprotinin, 10 μg/ml leupetin, 50 mM HEPES, pH 7.5). Crude lysates were sonicated and centrifuged at 10 000 rpm for 5 min at 4°C. Total protein concentration from the resulting supernatant was determined by bicinchoninic acid protein assay (Pierce, Rockford, IL) and normalized to 1 mg/ml. Aliquots of whole cell lysates were dried in a SpeedVac Concentrator (Savant, Holbrook, NY), denatured in Laemmli sample buffer, and stored at -20°C until use. Samples were boiled for 10 min, and 30 μg protein per lane was resolved on SDS-PAGE before being transferred to PVDF membranes. Membranes were incubated at 4°C overnight with appropriate primary antibodies (rabbit polyclonal anti-PKD, anti-p-PKD, anti-MEF2D, anti-phosphorylated MEF2D diluted 1:1000 (Cell Signaling Technology [Cambridge, MA]), and anti-ERK5, anti-phosphorylated ERK5 (p-ERK5) diluted 1:800 (Cayman Chemicals, Ann Arbor). After washing, the membrane was incubated with the corresponding horseradish peroxidase-conjugated secondary antibody. Immunoreactive bands were visualized with the SuperSignal West Pico enhanced chemiluminescence kit (Pierce, Rockford, IL) according to the manufacturer's instructions. Densitometric quantification was performed using Image-Pro Plus software with β-actin, lamin B1, PKD, ERK5, or MEF2D for normalization.
Immunofluorescence stainingCells cultured on glass coverslips were washed with PBS and fixed in 2% paraformaldehyde for 30 min at room temperature, followed by two rinses in PBS. Monolayers were then permeabilized three times for 10 min each, with PBS supplemented with 0.1% (final concentration) Triton X-100 and then blocked twice in PBS with 0.2% BSA for 10 min each at room temperature. Cells were incubated with 1/100 anti-phospho-ERK5 or anti-α-actinin (69758S, Cell Signaling Technology [Cambridge, MA]) in blocking solution overnight at 4°C. Coverslips incubated with rabbit polyclonal IgG, instead of primary antiserum, served as negative controls. After three washes for 10 min each in PBS with 0.2% BSA, coverslips were incubated with goat-anti-rabbit secondary antibody for 30 min and washed three times in PBS with 0.2% BSA. Immunolabeled cells were counterstained with DAPI to detect cell nuclei, then the slides were mounted. Samples were analyzed by confocal immunofluorescence microscopy with a Zeiss LSM 510 system. To avoid interference between fluorescence signals, images were captured in multitracking mode.
Real-time quantitative polymerase chain reactionReal-time quantitative polymerase chain reaction (PCR) was used to measure the mRNA levels of atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP), which are biomarkers of cardiac hypertrophy, using a SYBR Green PCR kit (Qiagen, Inc., Valencia, CA), according to the manufacturer's protocol. The following primers were used for real-time PCR: 5′-CGT ATA CAG TGC GGT GTC CA-3′ and 5′-GGT TGA CTT CCC CAG TCC AG-3′ for ANP; 5′-AGC TGC TGG AGC TGA TAA GAG-3′ and 5′-CTG CCC AAA GCA GCT TGA AC-3′ for BNP; and 5′-AAG AAG GTG GTG AAG CAG GC-3′ and 5′-TCC ACC ACC CTG TTG CTG TA-3′ for GAPDH. Amplification and detection were performed with the ABI 7500 Sequence Detection System (Applied Biosystems, Foster City, CA). The final real-time PCR analysis results are presented as the ratio of the mRNA of interest to that of GAPDH, which was used as an internal control. The relative mRNA levels were calculated using the 2-△△Ct method.
Small interfering RNA and its transfectionThe PKCɛ-specific small interfering RNA (siRNA) sequences used were as follows: #1, 5′-AAGATCGAGCTGGCTGTCTTT-3′; #2, 5′-TTCTAGCTCGACCGACAGAAA-3′; PKD: #1, 5′-GGAGGGCGAUCUUAUUGAATT- 3′; #2, 5′-CAGCAAACGUAGUGUAUUATT-3′; #3, 5′-CGCUAUAUUACCCAUGAAATT-3′; ERK5: #1, 5′-CAUGAACCCUGCCGAUAUUUU-3′; #2, 5′-AAACCAGUCUUUCGACAUGUU-3′; #3, 5′-GAACUGUGAGCUCAAGAUUUU-3′. They were obtained from Genepharma (Shanghai, China). Proliferating cells were removed by trypsinization, seeded into 60-mm dishes, and incubated for 24 h until they reached 60% confluence. Transfection of cardiomyocytes was achieved using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol, with 100 nM non-targeting control siRNA or SMART pool siRNA for PKCɛ, PKD and ERK5. After 48 h, cells were exposed to Ang II as indicated, and lysates underwent Western blot analysis.
Measurement of cell diameter and [3H]-leucine incorporationTo determine cell hypertrophy, cell diameter and [3H]-leucine (Leu) incorporation were measured. After being digested, centrifuged and resuspended in MEM, cells were counted in at least three dishes at each time point by phase-contrast microscopy. Cell diameter was measured using a digital photography analysis system. Ten fields were randomly chosen for each group, and 10 cardiomyocytes were determined for each field.
[3H]-Leu incorporation in cardiomyocytes was assessed as described previously. Cardiomyocytes were made quiescent by incubation in serum-free MEM for 24 h. Cells were incubated with 1 μCi/ml [3H]-Leu in the presence or absence of 100 nM Ang II. After 24 h, cells were washed twice with ice-cold PBS, and the proteins were precipitated in 5% trichloroacetic acid. [3H]-Leu incorporation was determined using a scintillation counter.
Statistical analysisThe experimental results are expressed as the mean ± standard error of the mean of three or more independent experiments and analyzed by one-way ANOVA with post-hoc comparisons using the LSD test. All statistical analyses were performed using IBM SPSS 22.0 statistical software (IBM SPSS, Inc., Chicago, IL). A p-value of <0.05 was considered statistically significant.
ResultsAngiotensin II induces PKCɛ-dependent PKD phosphorylation in cardiomyocytesIn accordance with our previous experiments, we selected a concentration of 100 nm of Ang II.13 Ang II (100 nmol/l) rapidly induced phosphorylation of PKD, within 5 min, with peak phosphorylation between 15 and 30 min, returning to baseline after 60 min (Figure 1A), which resembles the pattern of ERK5 phosphorylation in our previous experiments.13 During the course of Ang II stimulation, there was no obvious change in PKD expression, and β-actin levels showed equal loading in each sample. PKCɛ-specific siRNA significantly inhibited Ang II-induced activation of PKD (Figure 1B).
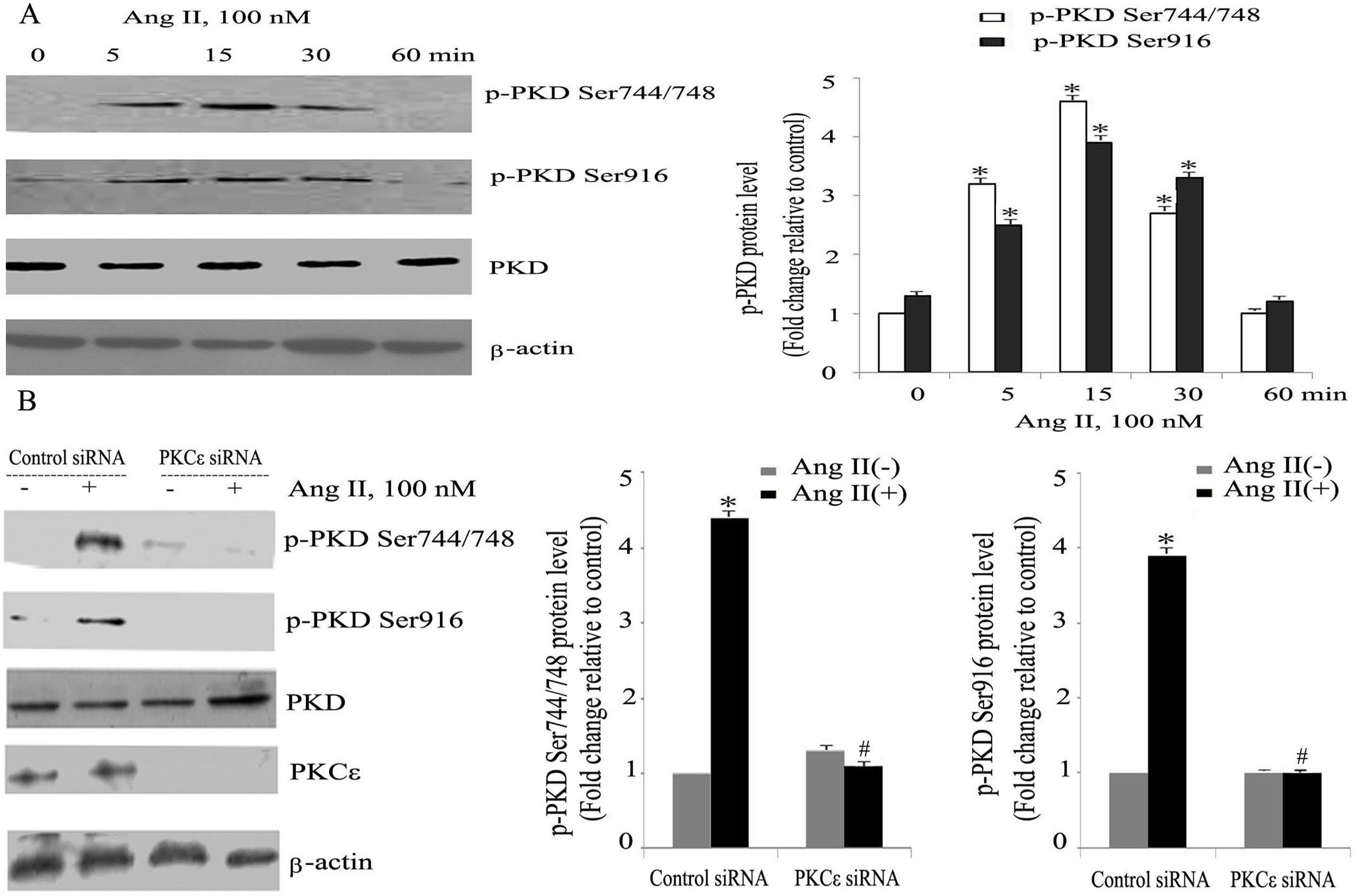
Angiotensin II (Ang II) stimulates PKCɛ-dependent PKD phosphorylation in cardiomyocytes. (A) Cardiomyocytes were stimulated with Ang II for various times. Phosphorylated PKD (p-PKD) protein levels were examined using Western blotting and normalized to the total levels of PKD. Ang II (100 nM, 0 min) was used as the control. Data are shown as mean ± standard error of the mean (SEM) of four separate experiments. *p<0.05 vs. controls; (B) cells were treated without or with Ang II (100 nM) and in the absence or presence of PKCɛ siRNA. Western blots (n=4) showed the phosphorylation of PKD and expression levels of PKCɛ and β-actin. Data are shown as mean ± SEM. *p<0.05 vs. control siRNA+ Ang II(-); #p<0.05 vs. control siRNA+Ang II(+).
To gain insight into the functional significance of PKD activation in Ang II-mediated signaling events, we determined whether knockdown of endogenous PKD in cardiomyocytes using siRNA affects Ang II-mediated ERK5 activation. Cardiomyocytes were transfected with PKD siRNA for 48 h and then stimulated with Ang II for the time indicated. Treatment of cardiomyocytes with PKD siRNA significantly reduced endogenous PKD expression, whereas control siRNA had no effect. PKD siRNA specifically targeted PKD since expression of β-actin and ERK5 was not changed. Decreasing PKD expression by its siRNA significantly inhibited Ang II-induced ERK5 activation in cardiomyocytes (Figure 2).
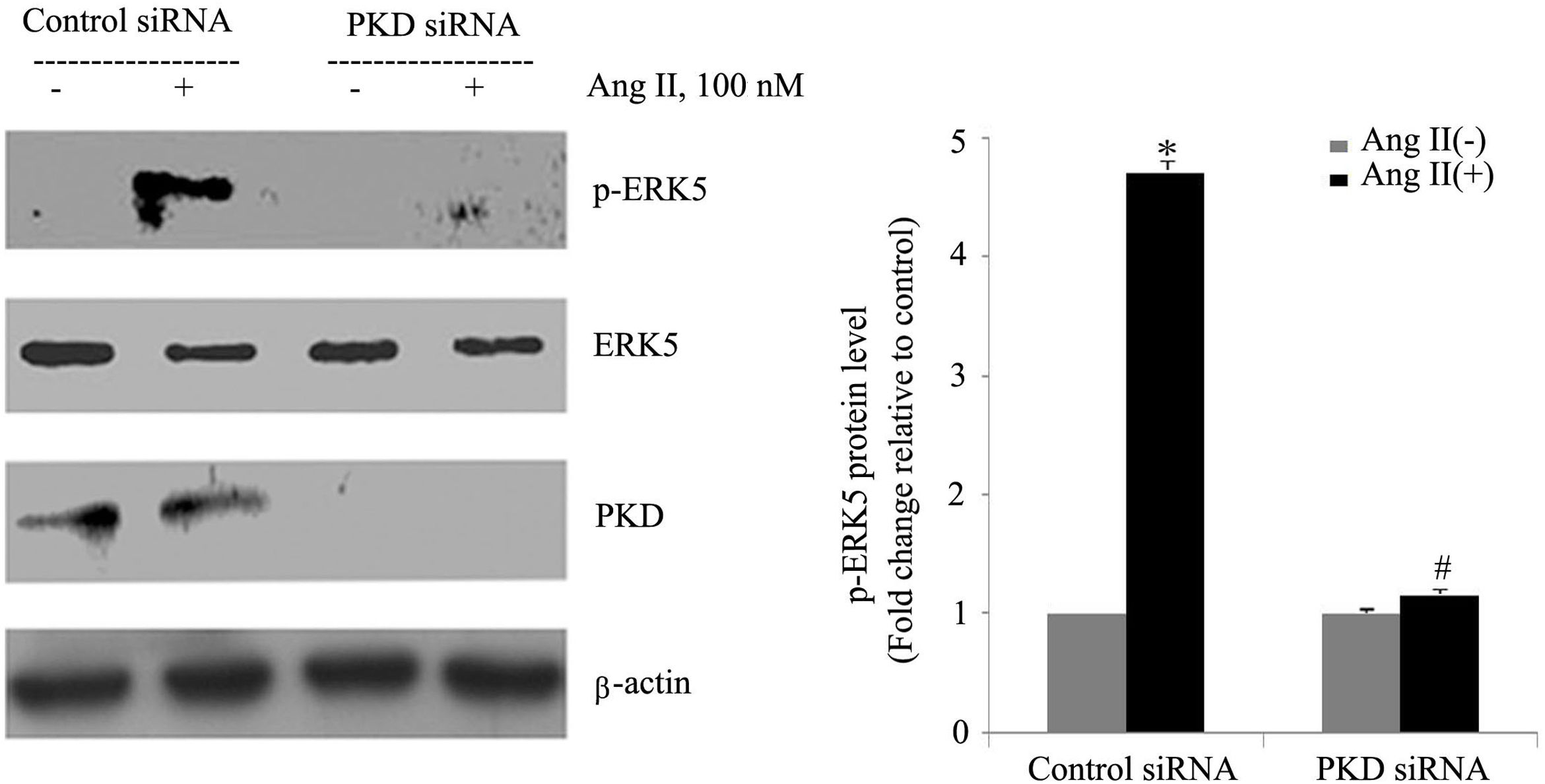
PKD specifically mediates angiotensin II (Ang II)-induced ERK5 phosphorylation. Cardiomyocytes were transfected with control or PKD small interfering RNA (siRNA), and then stimulated with Ang II. Phosphorylated ERK5 protein levels were examined using Western blotting and normalized to the total levels of ERK5. Data are shown as mean ± standard error of the mean of four separate experiments. *p<0.05 vs. control siRNA+ Ang II(-); #p<0.05 vs. control siRNA+Ang II(+).
Our previous studies showed that ERK5 was located primarily in the cytoplasm of cardiomyocytes before stimulation; nuclear entry was seen at 5 min after Ang II stimulation; marked translocation from the cytoplasm to the nucleus was observed by 15 min after addition of Ang II; and after 60 min of Ang II treatment, ERK5 was gradually shuttled back to the cytoplasm.13 To gain further insights into the signaling pathways leading to Ang II-induced translocation of ERK5, we studied the effects of PKD. Knocking down PKD expression by siRNA greatly attenuated Ang II-induced translocation of ERK5 in cardiomyocytes (Figure 3A).
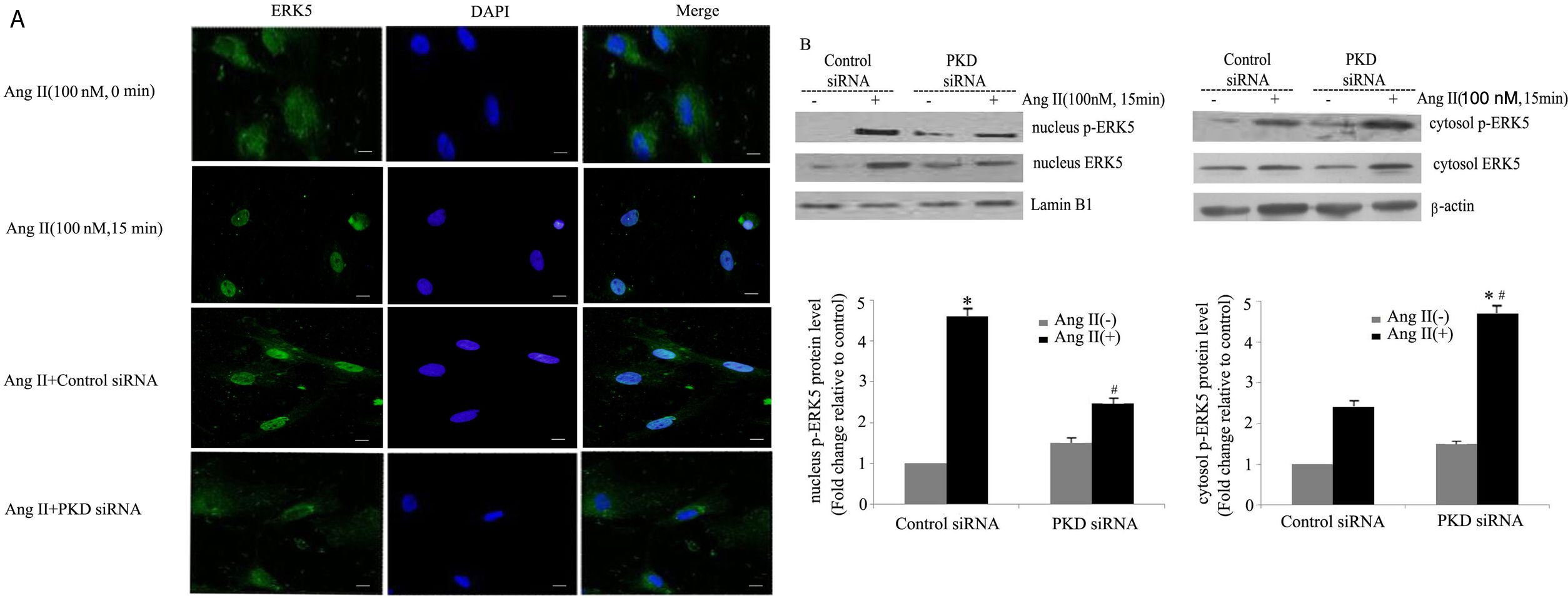
PKD specifically mediates angiotensin II (Ang II)-induced ERK5 translocation. (A) Immunofluorescence staining for ERK5, green (magnification 40×), scale bar 50 μm. Cardiomyocytes were transfected with control or PKD small interfering RNA (siRNA), and then stimulated with Ang II for 15 min. The representative images of fluorescence showed the subcellular localization of the proteins (n=4); (B) Western blot and quantitative analysis of p-ERK5 expression between the nucleus and cytoplasm due to Ang II (100 nM, 15 min) stimulation. Data are shown as the mean ± standard error of the mean of four separate experiments. *p<0.05 vs. control siRNA+ Ang II(-); #p<0.05 vs. control siRNA+Ang II(+).
We also demonstrated changes in the proportion of p-ERK5 content between the nucleus and cytoplasm. We found an increase in nuclear content of p-ERK5 and a concomitant decrease in cytoplasmic content under Ang II stimulation. Silencing PKD by siRNA significantly reduced the expression of ERK5 in the nucleus (Figure 3B).
Our previous studies confirmed that PKCɛ-mediated ERK5 translocation was involved in cardiomyocyte hypertrophy due to Ang II.13 Taken together, these results suggest that the PKCɛ/PKD pathway plays a part in the nucleocytoplasmic traffic of ERK5 in cardiomyocytes.
The PKCɛ/PKD/ERK5 pathway is involved in angiotensin II-induced activation of MEF2DTo determine whether the PKCɛ/PKD/ERK5 pathway plays an important role in Ang II-induced phosphorylation of MEF2D in cardiomyocytes, we examined the effect of PKCɛ, PKD and ERK5 siRNA on Ang II-induced MEF2D activation. Ang II induced phosphorylation of MEF2D after 5 min, with peak phosphorylation at 15 min, returning to baseline levels by 30 min (p<0.05) (Figure 4A). Knocking down PKCɛ, PKD and ERK5 expression by siRNA all significantly attenuated activation of MEF2D (Figure 4B).
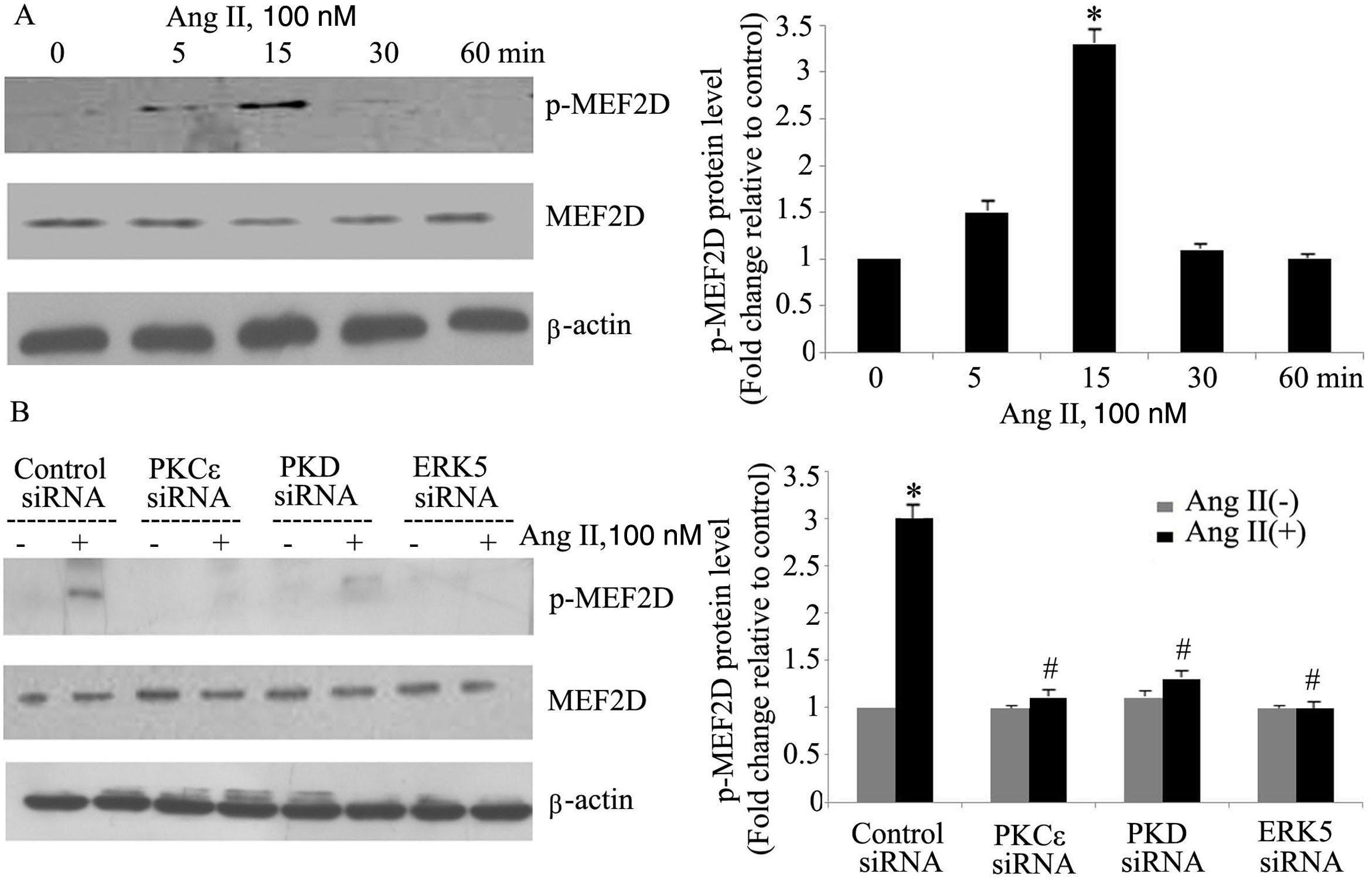
The PKCɛ/PKD/ERK5 pathway is involved in angiotensin II (Ang II)-induced MEF2D activation. (A) Cardiomyocytes were stimulated with Ang II for different times (0-60 min). Phosphorylated MEF2D protein levels were examined using Western blotting and normalized to the total levels of MEF2D. Data are shown as the mean ± standard error of the mean of four separate experiments. *p<0.05 vs. control without Ang II stimulation; (B) cardiomyocytes were treated without or with Ang II (100 nM) and in the absence or presence of PKCɛ, PKD or ERK5 small interfering RNA (siRNA). *p<0.05 vs. control siRNA+ Ang II(-); #p<0.05 vs. control siRNA+Ang II(+).
As the targets of MEF2D, ANP and BNP are biomarkers of cardiac hypertrophy. Compared with controls, Ang II significantly increased the mRNA expression of ANP and BNP, while PKCɛ, PKD and ERK5 siRNA treatment attenuated these increases (Figure 5).
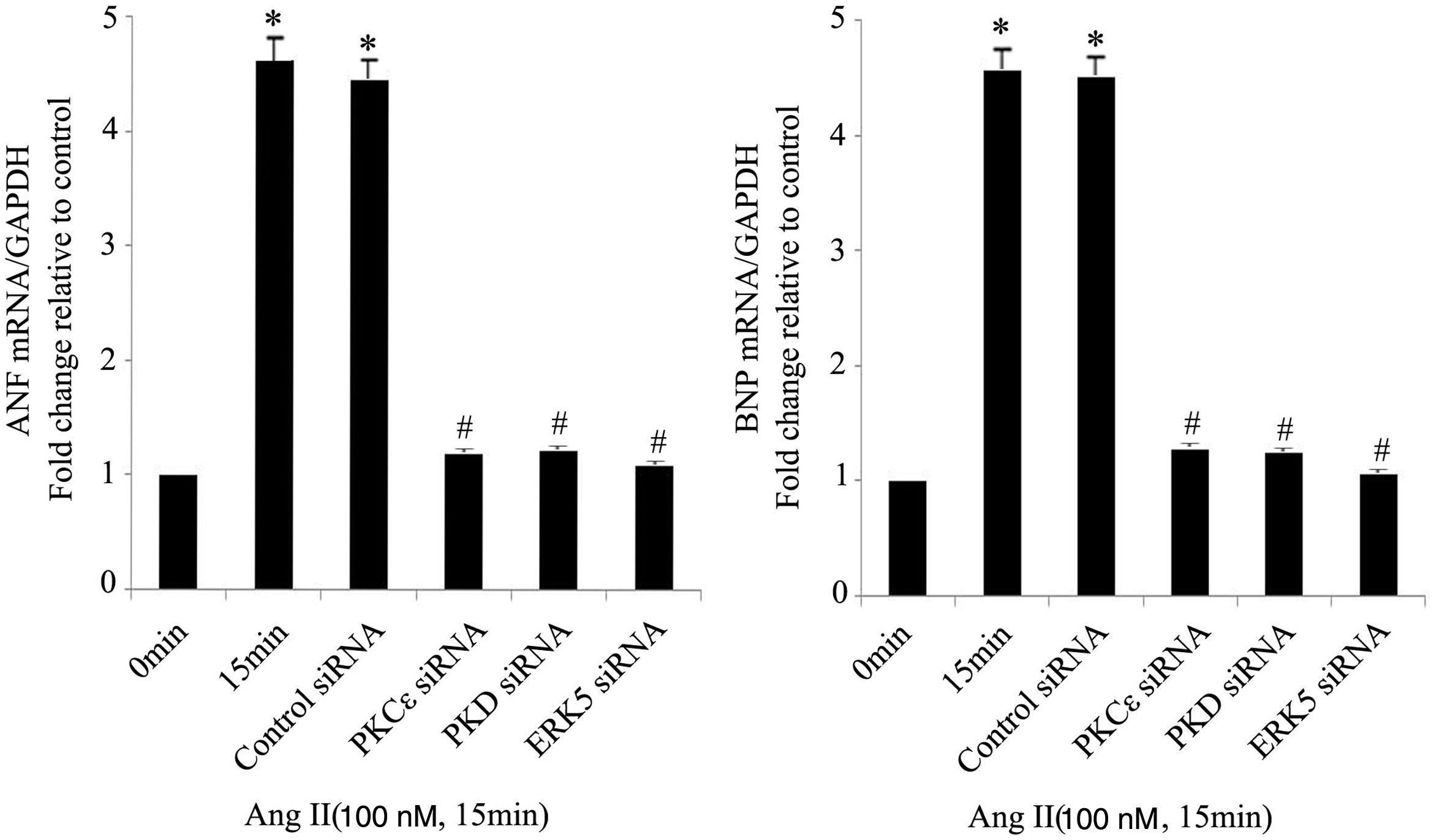
The PKCɛ/PKD/ERK5 pathway is involved in angiotensin II (Ang II)-induced expression of ANP and BNP mRNA. Cardiomyocytes were treated without or with Ang II (100 nM) and in the absence or presence of PKCɛ, PKD or ERK5 small interfering RNA (siRNA). ANP and BNP mRNA levels were examined using real-time quantitative polymerase chain reaction and normalized to GAPDH levels. Data are shown as mean ± standard error of the mean of four separate experiments. *p<0.05 vs. control (Ang II 0 min); #p<0.05 vs. control siRNA.
Both [3H]-Leu incorporation and cell size were significantly greater in Ang II-treated (100 nM) cells than in controls (p<0.05). Immunostaining for a cardiomyocyte marker (α-actinin) is included to attest the purity of the primary cultures (Figure 6A). Previous studies confirmed that the PKCɛ/ERK5 pathway participates in Ang II-induced cardiomyocyte hypertrophy. In this study, we confirmed the involvement of PKD in this process. As shown in Figure 6B and C, Ang II promotes cardiomyocyte hypertrophy, while PKCɛ, PKD and ERK5 siRNA greatly inhibited hypertrophy. PKD siRNA strongly suppressed Ang II-induced [3H]-Leu incorporation (p<0.05). Moreover, Ang II treatment increased the cell size of cardiomyocytes, whereas PKCɛ, PKD and ERK5 siRNA markedly decreased cell size (Figure 6D). These results suggest that the PKCɛ/PKD/ERK5 pathway plays pivotal roles in Ang II-induced cardiomyocyte hypertrophy.
![The PKCɛ/PKD/ERK5 pathway is implicated in angiotensin II (Ang II)-induced cardiomyocyte hypertrophy. (A) Immunostaining for a cardiomyocyte marker (α-actinin); (B-D) cardiomyocytes were treated without or with Ang II (100 nM) and in the absence or presence of PKCɛ or PKD or ERK5 siRNA. Microscopy of myocardial cells (B) and [3H]-leucine uptake in cells were analyzed (C), cell size was also detected (D) (n=4). Scale bar 50 μm. Data are shown as the mean ± standard error of the mean. *p<0.05 vs. control siRNA +Ang II(-); #p<0.05 vs. control small interfering RNA (siRNA) +Ang II(+); +p<0.05 vs. control (Ang II 0 min); ++p<0.05 vs. control siRNA.](https://static.elsevier.es/multimedia/08702551/0000004000000003/v1_202103231637/S0870255120304650/v1_202103231637/en/main.assets/gr6.jpeg?xkr=ue/ImdikoIMrsJoerZ+w94UphxYc+GPca8Z7OggvdfJQF4SIqTc4zp8SrbcUWBiKmVgt7PBOV8b/TxsdqahTQ+lt/f+rFfyXirMUg35S19lyLQlBwGm2BAqtYC9pkBOiTxbnPcnEuZmWTtCK91/YjEdO6LFV7X3gtm7SqkP5pY0J8LtfKo47ClSan+mzaGKpMxO4xW5+xdwJUwbfIvv7lnz8XeSQr1U0966oeL+9RfoSjiCKCtQITEl5VASwNvchyg3+EIofFmz9qni4kaqfKP1F0v4E5eZVUAsUQpB0uRk=)
The PKCɛ/PKD/ERK5 pathway is implicated in angiotensin II (Ang II)-induced cardiomyocyte hypertrophy. (A) Immunostaining for a cardiomyocyte marker (α-actinin); (B-D) cardiomyocytes were treated without or with Ang II (100 nM) and in the absence or presence of PKCɛ or PKD or ERK5 siRNA. Microscopy of myocardial cells (B) and [3H]-leucine uptake in cells were analyzed (C), cell size was also detected (D) (n=4). Scale bar 50 μm. Data are shown as the mean ± standard error of the mean. *p<0.05 vs. control siRNA +Ang II(-); #p<0.05 vs. control small interfering RNA (siRNA) +Ang II(+); +p<0.05 vs. control (Ang II 0 min); ++p<0.05 vs. control siRNA.
Cardiac hypertrophy is an adaptive and compensatory mechanism preserving cardiac output during hypertension. Nevertheless, long-term stimuli incite chronic hypertrophy and may lead to heart failure. Inappropriate activation of the renin-angiotensin-aldosterone system (RAAS) plays an important role in this process. Ang II, the main effector of the RAAS, is increased in patients and animal models of hypertension and cardiac hypertrophy. Ang II can induce cardiomyocyte hypertrophy in vitro. A large number of intracellular pathways have been associated with this hypertrophic response.15 These pathways are intertwined, generating a complex response that is still not completely understood. In this study, we provide evidence that PKD activation is one of the early signaling events in cardiomyocytes in response to Ang II.
The experiments presented here were designed to elucidate the underlying mechanisms of cardiomyocyte hypertrophy by Ang II. In the present study, we demonstrated that Ang II stimulated PKD phosphorylation in a time-dependent manner in cardiomyocytes. PKCɛ is the specific upstream regulator of PKD, and inhibiting PKCɛ by siRNA reduced PKD activation. Furthermore, PKD is critical in ERK5 phosphorylation and translocation, since silencing PKD by siRNA blocked ERK5 activation and nucleocytoplasmic traffic. Importantly, the PKCɛ/PKD/ERK5 pathway plays a significant role in MEF2D activation and ANP and BNP mRNA expression, and contributes to Ang II-induced cardiomyocyte hypertrophy, as shown by significantly increased [3H]-Leu incorporation. This is the first report to show the critical role of the PKCɛ/PKD/ERK5/MEF2D pathway in cardiomyocyte hypertrophy, which may provide new insights into the molecular mechanisms of cardiac remodeling observed in animal models and in humans.
PKCɛ is activated in response to hypertrophic stimuli in cultured myocytes and in vivo; overexpression and activation of PKCɛ results in myocardial hypertrophy.16 Cardiac-specific transgenic mice overexpressing a constitutively active PKCɛ mutant developed cardiac hypertrophy.17 Our previous studies found involvement of PKCɛ in Ang II-induced cardiomyocyte hypertrophy. Knocking down PKCɛ by specific siRNA strongly blocked Ang II-stimulated activation and translocation of ERK5 as well as [3H]-Leu incorporation in cardiomyocytes.13
PKD lies downstream of PKCs in a novel signal transduction pathway implicated in the regulation of multiple fundamental biological processes. In recent years, PKD has been implicated as a signal transducer in cardiac hypertrophy.18 Knockdown of PKD expression with siRNA blunts agonist-dependent hypertrophy, whereas in vivo cardiac-specific expression of constitutively active PKD causes a brief phase of cardiac hypertrophy.14 Specific cellular processes that appear to be under PKD control include transcriptional factor activation.19 Activated PKD, perhaps along with other histone deacytelase (HDAC) kinases, primarily promotes MEF2 expression and activation by inducing class II HDAC phosphorylation and nuclear export. A-kinase anchoring protein 13 (AKAP13) has been well documented as a vital component of the MEF2-mediated fetal gene response and the cardiac hypertrophic signaling cascade.20,21 AKAP13 binds PKD near the C-terminus of the protein,22 and upon activation by PKC, PKD dissociates from AKAP13 and phosphorylates HDAC5 in the nucleus, causing dissociation of HDAC5 from MEF2.22,23
The MEF2 family has four members: MEF2A, MEF2B, MEF2C and MEF2D. There is increasing evidence that MEF2C and MEF2D both act as hypertrophy mediators. Expression of MEF2C was increased in both cardiomyocytes and human aortic smooth muscle cells (HASMCs) in Ang II-induced hypertrophy.13,24 MEF2D-null mice were resistant to cardiac hypertrophy in response to pressure overload and chronic adrenergic stimulation.25 Our previous studies also confirmed that PKD participates in cardiac hypertrophy by regulating MEF2D in spontaneously hypertensive rats.26 Consistent with this, our study showed that Ang II induces PKCɛ/PKD-dependent MEF2D activation and cardiomyocyte hypertrophy. Inhibiting PKCɛ or PKD by specific siRNA strongly reduced activation of MEF2D.
It has been demonstrated that ERK5 is involved in the development of detrimental hypertrophy in different pathological systems.27 Mice with cardiomyocyte-specific deletion of ERK5 showed a reduced hypertrophic response during pressure overload.28 ERK5 is closely associated with the activation of MEF2 transcription factors.29 In sensory neurons, the ERK5/MEF2D pathway strictly regulates expression of Bcl-w, an anti-apoptopic bcl-2 family member, which promotes sensory neuron survival.30 A groundbreaking finding revealed that activation of MEF2C/D transcription factors promotes early B-cell development, which depends on their phosphorylation by ERK5.31 In the present study, we ascertained that Ang II-induced ERK5 phosphorylation resulted in MEF2D activation and subsequent cardiomyocyte hypertrophy. Knocking down ERK5 by siRNA significantly attenuated MEF2D activation and [3H]-Leu incorporation.
The subcellular location of ERK5 was cell type-specific.32,33 Recently, a mechanism for the nucleocytoplasmic transport of ERK5 has been proposed. ERK5 has nuclear localizing activity in its C-terminal region, owing to a bipartite nuclear localization signal.33,34 In addition, ERK5 has nuclear export activity.35 The balance between nuclear import and export determines the subcellular localization of ERK5.34 A previous study showed that ERK5 is mainly cytosolic in quiescent cardiomyocytes, but translocates to the nucleus during activation by Ang II. PKCɛ is critical for ERK5 nuclear entry.13
Our previous studies showed that the PKD/ERK5 pathway plays an important role in Ang II-induced hypertrophy of HASMCs.24 But the effect of PKD on ERK5 in cardiomyocytes was still unclear. Since they have a similar activation pattern and common upstream and downstream molecules, we speculate that PKD plays a critical role in ERK5 phosphorylation and translocation in cardiomyocytes by Ang II. In agreement with this, the present study found that knocking down PKD by siRNA attenuated Ang II-stimulated ERK5 phosphorylation, followed by decreased translocation.
ConclusionIn summary, we demonstrate for the first time that Ang II induces PKD-dependent ERK5 phosphorylation and translocation in cardiomyocytes. The PKCɛ/PKD/ERK5 pathway also plays an essential role in Ang II-stimulated MEF2D activation and cardiomyocyte hypertrophy. These results provide new insights into the molecular underpinnings of cardiomyocyte hypertrophy in response to Ang II. These findings may provide novel targets for therapeutic manipulation through pharmacological or genetic approaches to pathological cardiac remodeling in hypertension.
Conflicts of interestThe authors have no conflicts of interest to declare.
This work was supported by the National Natural Science Foundation of China (no. 81670245). We are very grateful for Dr. Zhuo Zhao, who is the leader of this project.
