







This study aims to assess the long-term survival of pulmonary arterial hypertension (PAH) and chronic thromboembolic pulmonary hypertension (CTEPH) patients followed in a Portuguese pulmonary hypertension (PH) referral center.
MethodsWe studied PAH and CTEPH patients diagnosed between January 2005 and December 2016. Cumulative survival was estimated using the Kaplan-Meier method. Survival trends were compared over two periods (2005-2010 vs. 2011-2016).
ResultsOf the 142 studied PH patients (age 54±18 years; 31% male), 47 had CTEPH and 95 had group 1 PH. Most patients with CTEPH and idiopathic/heritable PAH (I/HPAH) were in NYHA III-IV at diagnosis (64% and 57%, respectively). At the time of death, 31% of patients with connective tissue disease (CTD)-associated PAH (CTD-PAH) and all I/HPAH patients were on double or triple combination therapy. No patient underwent lung transplantation. Pulmonary endarterectomy or angioplasty were performed in 36% of CTEPH patients. Age at diagnosis tended to increase over time in CTD-PAH (53±15 vs. 63±15 years; p=0.13) and I/HPAH (39±15 vs. 51±19 years; p=0.10). The five-year survival estimates for I/HPAH, CTD-PAH and CTEPH patients were 80%, 52%, and 81%, respectively. Over time, CTD-PAH and CTEPH showed better five-year survival (33 vs. 67% and 77 vs. 84%), but I/HPAH did not (84 vs. 75%).
ConclusionsOur data indicate a trend toward improved survival over time of CTD-PAH and CTEPH patients treated at a Portuguese referral PH center. Earlier diagnosis, increasing use of parenteral prostanoids, and surgical treatment may further improve PH prognosis.
Este estudo tem como objetivo avaliar a sobrevivência a longo prazo em doentes com hipertensão arterial pulmonar (PAH) e hipertensão pulmonar tromboembólica crónica (CTEPH) seguidos num centro de tratamento.
MétodosDoentes diagnosticados com PAH ou CTEPH entre janeiro 2005 e dezembro 2016 foram incluídos. A sobrevivência cumulativa foi estimada utilizando o método Kaplan-Meier. Compararam-se os padrões de sobrevivência de dois períodos (2005-2010 versus 2011-2016).
ResultadosForam estudados 142 doentes (54 ± 18 anos; 31% homens), 47 diagnosticados com CTEPH e 95 com PAH. A maioria dos doentes com CTEPH e etiologia idiopática/hereditária (I/HPAH) apresentava classe funcional NYHA III-IV ao diagnóstico (64% e 57%, respetivamente). Aquando da morte, 31% dos doentes com doença do tecido conjuntivo (CTD) e todos os doentes com H/IPAH recebiam terapia dupla ou tripla. Nenhum doente realizou transplante pulmonar. Endarterectomia pulmonar (PEA) ou angioplastia foram realizadas em 36% dos doentes com CTEPH. A idade de diagnóstico de H/IPAH (39 ± 15 versus 51 ± 19 anos; p = 0,10) e CTD (53 ± 15 versus 63 ± 15 anos; p = 0.13) tendeu a aumentar. A sobrevivência a cinco anos foi estimada em 80%, 52% e 81% para H/IPAH, CTD e CTEPH, respetivamente. No 2.° período, a sobrevivência a cinco anos melhorou nos doentes com CTD e CTEPH (33% versus 67% e 77% versus 84%), mas não nos I/HPAH (84% versus 75%).
ConclusõesExiste uma tendência de melhoria na sobrevivência de doentes com CTD-PAH e CTEPH tratados num centro de referência português. O diagnóstico precoce, o uso de prostanoides parenterais e a disponibilização de tratamentos cirúrgicos poderão traduzir-se em ganhos adicionais de sobrevida.
Pulmonary hypertension (PH) is characterized by an increase in pulmonary artery pressure. It can be associated with a wide range of conditions,1 the most common of which are left heart disease and lung disease, in which PH has considerable prognostic significance but no indication for specific therapy.2 The distinction between these causes and less frequent causes of PH such as pulmonary arterial hypertension (PAH) and chronic thromboembolic pulmonary hypertension (CTEPH) is often challenging and always critical, since the latter have an ominous prognosis without specific treatment.3,4 Ten specific pulmonary vasodilators are currently available for PAH and there is robust evidence on the clinical benefits of pulmonary endarterectomy (PEA) for CTEPH patients.2
Despite notable advances in recent decades, rare diseases such as PAH and CTEPH still present numerous challenges. Early diagnosis is fundamental but still an unmet need. The more advanced the functional class at diagnosis, the worse the prognosis.5,6 Regarding treatment, generalization of data from clinical trials is not straightforward, as these trials’ study populations often differ from real-world patients.7 Observational data from registries and cohort studies are accordingly crucial to provide insights on epidemiology, adherence to guidelines, effectiveness of treatments, and outcomes in clinical practice. Several international registries and PH referral center cohort studies have been published.8 However, available data on PAH and CTEPH patients followed in Portugal are limited, especially in terms of long-term mortality.9–11
We studied PAH and CTEPH patients followed in a Portuguese PH referral center with the following aims: (1) to describe patients’ clinical, functional and hemodynamic profile at diagnosis; (2) to characterize the use of specific pulmonary vasodilator treatments; and (3) to examine long-term mortality and survival trends between different time periods.
MethodsThis retrospective single-center study included all group 1 and group 4 PH patients followed in the Pulmonary Vascular Disease Unit of Hospital de Santo António, Centro Hospitalar do Porto (Porto, Portugal) between January 2005 and December 2016. This unit is the official referral center for the treatment of PH in the Northern region of the country, covering an adult population of 3.8 million. Demographic, clinical, hemodynamic, and treatment data were collected using dedicated PH software (PAHTool®, Inovultus Lda., Santa Maria da Feira, Portugal). Specific vasodilator treatments were collected from the last follow-up observation. All patients had incident PAH confirmed by right heart catheterization (RHC) using the criteria and PH classification from current international clinical guidelines.2 The ethics committee of Hospital de Santo António authorized the study and waived patients’ consent. Survival status (all-cause mortality) was assessed by chart review through February 1, 2017.
Demographic and clinical variables were summarized with descriptive statistics. Categorical variables were summarized as absolute frequency and percentage, and continuous variables were summarized as mean and standard deviation. Bivariate analysis was conducted using the Student's t test, Wilcoxon's test, Fisher's exact test, or chi-square tests, as appropriate. Cumulative survival was estimated using the Kaplan-Meier method. Multivariate Cox proportional hazards regression models were used to assess the unadjusted and adjusted association of PH etiology and mortality. To describe survival trends over time, patients diagnosed in two different six-year periods (2005-2010 vs. 2011-2016) were compared. A 5% significance level was employed for all analyses. The statistical analysis was performed using Stata software, version 12.1 (StataCorp LP, College Station, TX, USA).
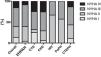
ResultsOverall populationWe studied 47 group 4 (CTEPH) and 95 group 1 PH (PAH) patients. The latter had the following etiologies: congenital heart disease (CHD; n=32), idiopathic/heritable (I/HPAH; n=25), connective tissue disease (CTD; n=28), human immunodeficiency virus infection (n=3) and portal hypertension (n=7). The clinical characteristics of the overall study population are described in Table 1. Most group 1 and 4 PH patients were women and were in an advanced functional class (New York Heart Association [NYHA] III or IV) at diagnosis (Figure 1). CTEPH patients were older and had higher N-terminal pro-brain natriuretic peptide (NT-proBNP) levels despite lower mean pulmonary artery pressures.
Clinical characteristics of the study population at diagnosis.
| Overall (n=142) | PAH (n=95) | CTEPH (n=47) | p | |
|---|---|---|---|---|
| Age, years | 54±18 | 49±18 | 64±14 | <0.001 |
| Male, n (%) | 44 (31) | 32 (34) | 12 (26) | 0.32 |
| Diagnosis, n (%) | ||||
| I/HPAH | 25 (18) | 25 (26) | NA | |
| CTD | 28 (20) | 28 (29) | NA | |
| CHD | 32 (23) | 32 (34) | NA | |
| PoPH | 7 (5) | 7 (7) | NA | |
| HIV | 3 (2) | 3 (3) | NA | |
| NYHA, n (%) | 0.41 | |||
| I/II | 52 (38) | 33 (36) | 19 (43) | |
| III/IV | 84 (62) | 59 (64) | 25 (57) | |
| 6MWT, m | 313±134 | 307±140 | 327±119 | 0.46 |
| NT-proBNP, pg/ml | 741 (253-2202) | 570 (207-2183) | 1071 (363-2657) | 0.04 |
| Hemodynamics | ||||
| mPAP, mmHg | 51±16 | 53±18 | 45±7 | 0.03 |
| RAP, mmHg | 8±5 | 8±4 | 9±6 | 0.71 |
| CI, l/min/m2 | 2.8±0.8 | 2.9±0.9 | 2.7±0.6 | 0.39 |
| PVR, WU | 9±5 | 9±5 | 8±3 | 0.52 |
6MWT: 6-min walk test; CHD: congenital heart disease; CI: cardiac index; CTD: connective tissue disease; CTEPH: chronic thromboembolic pulmonary hypertension; HIV: pulmonary hypertension associated with human immunodeficiency virus infection; I/HPAH: idiopathic/heritable pulmonary arterial hypertension; mPAP: mean pulmonary artery pressure; NA: not applicable; NT-proBNP: N-terminal pro-brain natriuretic peptide; NYHA: New York Heart Association functional class; PAH: pulmonary arterial hypertension; PoPH: portopulmonary hypertension; PVR: pulmonary vascular resistance; RAP: right atrial pressure; WU: Wood units. NT-proBNP levels are presented as median and interquartile range.
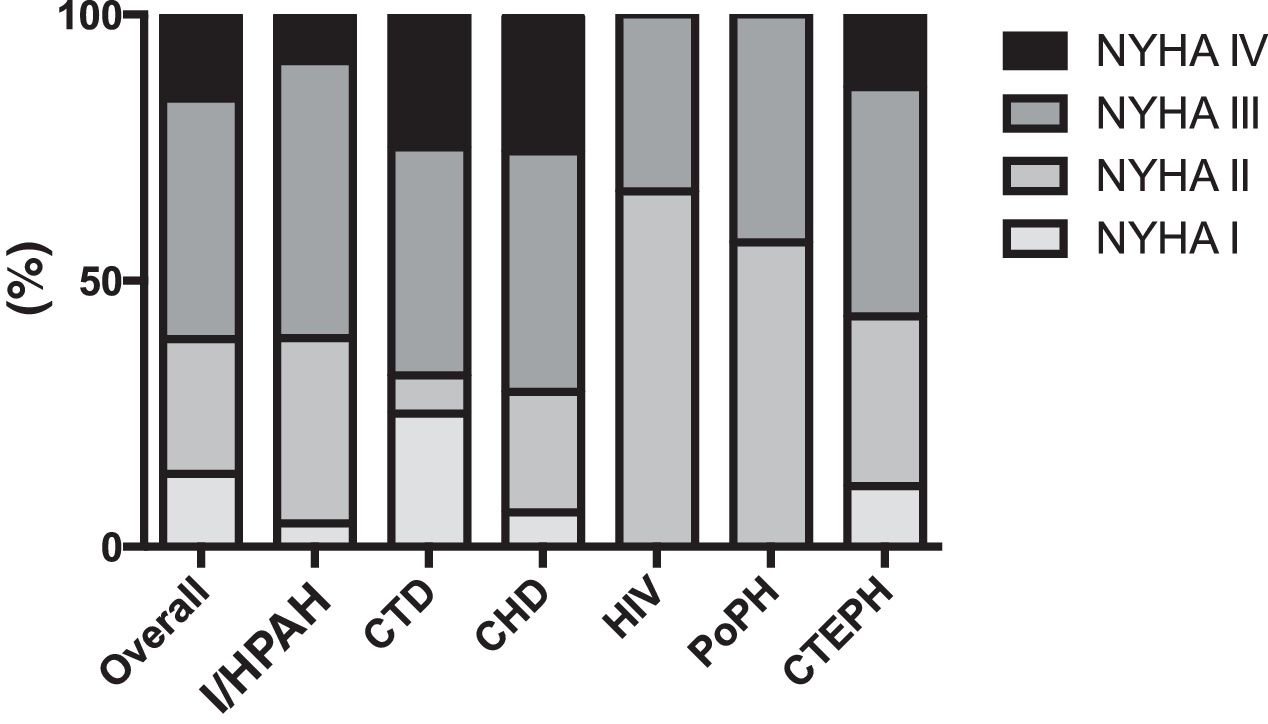
Functional capacity measured by NYHA functional class at time of diagnosis of pulmonary hypertension patients according to etiology. CHD: congenital heart disease; CTD: connective tissue disease; CTEPH: chronic thromboembolic pulmonary hypertension; HIV: pulmonary hypertension associated with HIV infection; I/HPAH: idiopathic/heritable pulmonary arterial hypertension; NYHA: New York Heart Association functional class; PoPH: portopulmonary hypertension.
Fifty-three percent of PAH patients were on two or three vasodilators, while 21% of CTEPH patients were on combination therapy (Table 2). Thirty-six percent of CTEPH patients received non-pharmacological treatment, either endarterectomy (n=14) or angioplasty (n=3). Reasons for CTEPH patients not undergoing interventional therapy were patient refusal in 20 (43%) and considered inoperable or at high operative risk in 10 (21%). None of the patients underwent lung transplantation.
Pulmonary hypertension treatment at last follow-up visit.
| Overall (n=142) | PAH (n=95) | CTEPH (n=47) | |
|---|---|---|---|
| Pharmacological class, n (%) | |||
| CCB | 6 (4) | 6 (6) | 0 (0) |
| ERA | 105 (74) | 84 (88) | 21 (45) |
| PDE5i | 59 (42) | 51 (54) | 8 (17) |
| sGC | 13 (9) | 0 (0) | 13 (28) |
| Prostanoids | 20 (14) | 19 (20) | 1 (2) |
| Combination therapy, n (%) | |||
| Single | 60 (42) | 38 (40) | 22 (47) |
| Double | 43 (30) | 34 (36) | 9 (19) |
| Triple | 17 (12) | 16 (17) | 1 (2) |
| Non-pharmacological treatment, n (%) | |||
| PEA/pulmonary angioplasty | 17 (12) | 0 (0) | 17 (36) |
CCB: calcium channel blockers; CTEPH: chronic thromboembolic pulmonary hypertension; ERA: endothelin-1 receptor antagonists; PAH: pulmonary arterial hypertension; PDE5i: phosphodiesterase-5 inhibitors; PEA: pulmonary endarterectomy; sGC: soluble guanylate cyclase.
The clinical characteristics of patients diagnosed in the periods 2005-2010 and 2011-2016 are displayed in Table 3. Over time, age at diagnosis tended to increase in CTD-associated PAH (CTD-PAH) and I/HPAH patients and decreased significantly in CHD. At diagnosis, most patients with CTD and I/HPAH were in NYHA functional class III-IV, a scenario that did not improve over time. By contrast, CTEPH patients were predominantly in NYHA I-II at diagnosis in the more recent period (2011-2016). No significant hemodynamic differences at diagnosis were observed over time across the PH classification subgroups. In the I/HPAH subgroup, five (36%) patients diagnosed during the period 2005-2010 were positive on vasoreactivity testing, compared to only one (9%) during the period 2011-2016.
Clinical characteristics of patients diagnosed in the two different periods.
| I/H PAH | CTD-PAH | CHD PAH | CTEPH | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2005-2010 (n=14) | 2011-2016 (n=11) | p | 2005-2010 (n=9) | 2011-2016 (n=19) | p | 2005-2010 (n=15) | 2011-2016 (n=17) | p | 2005-2010 (n=13) | 2011-2016 (n=34) | p | |
| Age, years | 39±15 | 51±19 | 0.10 | 53±15 | 63±15 | 0.13 | 52±16 | 34±13 | 0.001 | 63±12 | 65±15 | 0.70 |
| Male, n (%) | 5 (36) | 4 (36) | 0.83 | 0 (0) | 3 (16) | 0.21 | 5 (33) | 11 (65) | 0.26 | 2 (15) | 10 (29) | 0.32 |
| NYHA, n (%) | 0.55 | 0.93 | 0.06 | 0.004 | ||||||||
| I/II | 6 (46) | 3 (30) | 3 (33) | 6 (32) | 2 (13) | 7 (44) | 1 (8) | 18 (56) | ||||
| III/IV | 7 (54) | 7 (70) | 6 (67) | 13 (68) | 13 (87) | 9 (56) | 11 (92) | 14 (44) | ||||
| 6MWT, m | 344±130 | 260±155 | 0.18 | 331±155 | 242±152 | 0.20 | 348±124 | 294±134 | 0.29 | 284±139 | 349±103 | 0.13 |
| NT-proBNP, pg/ml | 983 (169-3027) | 801 (375-1489) | 0.69 | 269 (204-2813) | 1297 (313-2277) | 0.67 | 1415 (534-2815) | 253 (58-376) | 0.001 | 2468 (556-4922) | 1049 (329-1905) | 0.14 |
| Hemodynamics | ||||||||||||
| mPAP, mmHg | 54±15 | 55±20 | 0.86 | 40±14 | 44±12 | 0.88 | 57±19 | 77±19 | 0.12 | 52±5 | 44±7 | 0.05 |
| RAP, mmHg | 10±5 | 11±6 | 0.51 | 6±3 | 6±2 | 0.91 | 8±4 | 6±3 | 0.32 | 9±4 | 9±6 | 0.81 |
| CI, l/min/m2 | 2.8±0.5 | 2.9±0.9 | 0.88 | 2.6±0.3 | 3.1±0.8 | 0.41 | 2.4±1.6 | 2.8±1.0 | 0.73 | 3.0±1.1 | 2.7±0.6 | 0.51 |
| PVR, WU | 12±8 | 9±4 | 0.42 | 6±4 | 7±4 | 0.72 | 11±4 | 14±7 | 0.47 | 9±4 | 8±3 | 0.81 |
| Medical therapya | 0.12 | 0.29 | 0.52 | 0.34 | ||||||||
| CCB, n (%) | 5 (36) | 1 (9) | 0.12 | 0 | 0 | 0 | 0 | 0 | 0 | |||
| Single, n (%) | 3 (21) | 3 (27) | 7 (78) | 8 (42) | 6 (40) | 6 (35) | 7 (54) | 15 (44) | ||||
| Double, n (%) | 2 (14) | 5 (46) | 1 (11) | 8 (42) | 7 (47) | 8 (47) | 4 (31) | 5 (15) | ||||
| Triple, n (%) | 7 (50) | 3 (27) | 1 (11) | 2 (11) | 2 (13) | 1 (6) | 0 (0) | 1 (3) | ||||
| Non-pharmacological treatment | ||||||||||||
| PEA/pulmonary angioplasty | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 5 (39) | 12 (35) | 0.84 | |||
Single, double and triple refer to specific pulmonary vasodilators (phosphodiesterase-5 inhibitors, endothelin-1 receptor antagonists, soluble guanylate cyclase and prostanoids).
6MWT: 6-min walk test; CCB: calcium channel blockers; CHD: congenital heart disease; CI: cardiac index; CTD: connective tissue disease; CTEPH: chronic thromboembolic pulmonary hypertension; I/HPAH: idiopathic/heritable pulmonary arterial hypertension; mPAP: mean pulmonary artery pressure; NT-proBNP: N-terminal pro-brain natriuretic peptide; NYHA: New York Heart Association functional class; PAH: pulmonary arterial hypertension; PVR: pulmonary vascular resistance; RAP: right atrial pressure; WU: Wood units. NT-proBNP levels are presented as median and interquartile range.
The trend toward higher percentages of triple combination therapy in the period 2005-2010 (Table 3) is related to the different follow-up time. At two-year follow-up, therapeutic aggressiveness in PAH patients was similar between periods (p=0.93), as 39% and 9% underwent double and triple combination therapy, respectively, in the 2005-2010 period, compared to 36% and 9% in the more recent period.
Survival analysisThe mean follow-up period was 4.4±3.3 years. There were 49 (35%) deaths. The mean one-year, three-year and five-year survival of group 1 and group 4 PH patients are presented in Figure 2. CTD-PAH patients had the worst prognosis, with only 52% patients surviving five years after diagnosis (Figure 2). At the time of death, 31% of CTD-PAH and all I/HPAH patients were on double or triple vasodilator therapy, while 20% were under prostanoid therapy. CTEPH patients who did not undergo PEA showed worse prognosis after five years of follow-up, with 72% survival, compared to those who were surgically treated (94%). CTEPH patients treated with PEA were younger (55±11 vs. 67±15 years; p=0.09), presented with a lower 6-min walking distance at diagnosis (277±150 vs. 353±92 m; p=0.07) and had higher pulmonary vascular resistance (10±3 vs. 7±3 Wood units; p=0.03). No other relevant differences were found between these patients.
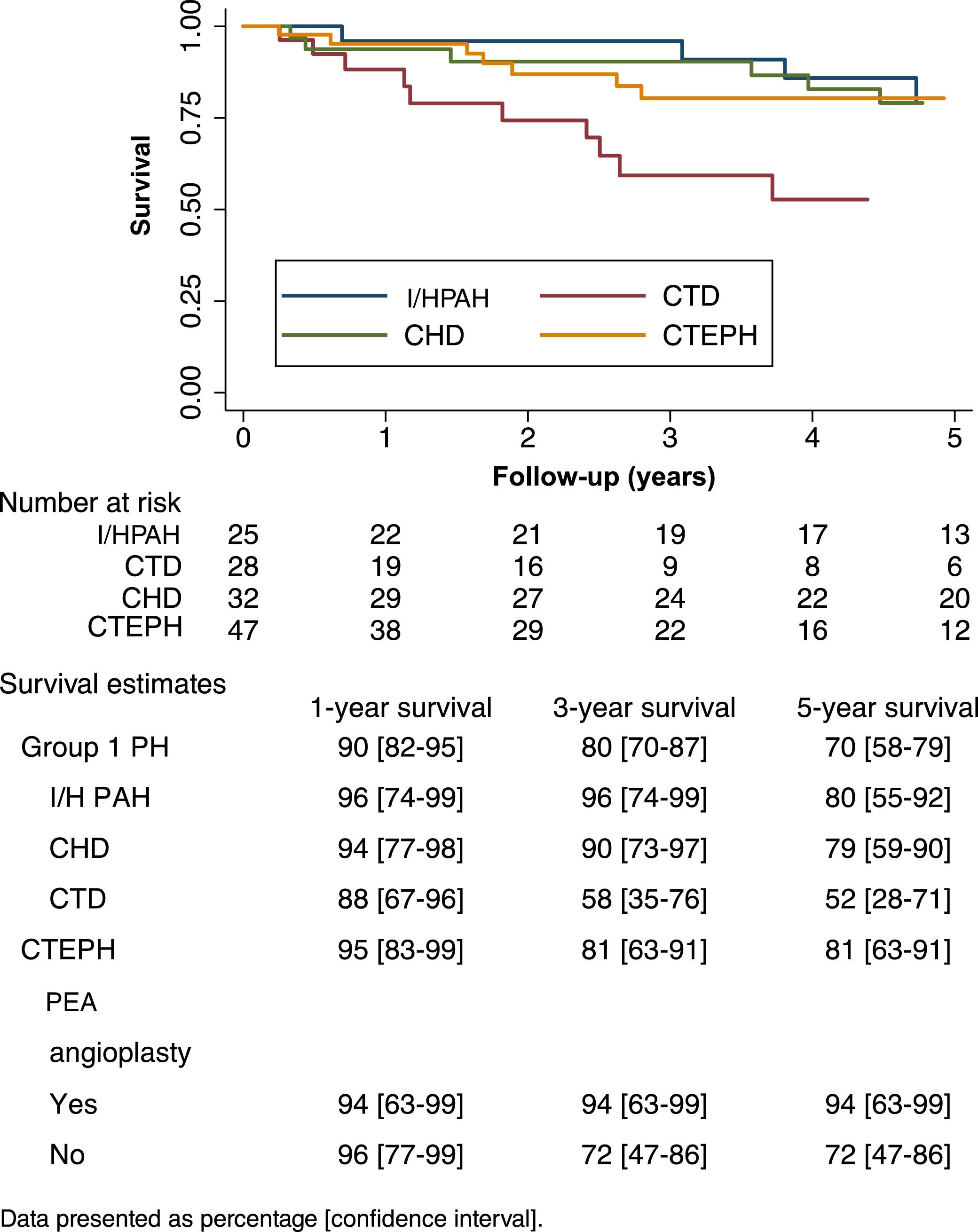
Survival curves of pulmonary hypertension patients according to etiology. CHD: congenital heart disease; CTD: connective tissue disease; CTEPH: chronic thromboembolic pulmonary hypertension; I/HPAH: idiopathic/heritable pulmonary arterial hypertension; PAH: pulmonary arterial hypertension; PEA: pulmonary endarterectomy; PH: pulmonary hypertension.
With regard to I/HPAH and CTD-PAH, while age (hazard ratio [HR] 1.08; 95% confidence interval [CI]: 1.04-1.16) and 6-min walking distance at diagnosis (HR 0.99; 95% CI: 0.99-0.99) were associated with prognosis, gender (HR 1.78; 95% CI: 0.70-4.53) and NYHA functional class (HR 2.06; 95% CI: 0.78-5.50) were not. Regarding CTEPH patients, in unadjusted analysis, PEA tended to be associated with lower mortality (HR 0.15; 95% CI: 0.02-1.22) but not after adjusting for age (HR 0.33; 95% CI: 0.04-2.97).
Figure 3 displays Kaplan-Meier curves and crude one-year, three-year and five-year survival estimates of I/HPAH, CTD-PAH and CTEPH patients diagnosed in the two different periods (2005-2010 and 2011-2016). The mean estimate of survival improved numerically over time in all etiologies, except for I/HPAH. The follow-up time (median and interquartile range) for patients included in 2005-2010 and 2011-2016 were 6.9 (3.7-9.5) years and 2.8 (1.2-4.8) years, respectively. The annual incidence rates in these periods were 9.3 (6.6-13.2) and 6.1 (3.8-9.7) per 100 person/years, respectively. Kaplan-Meier curves are shown in Figure 3.
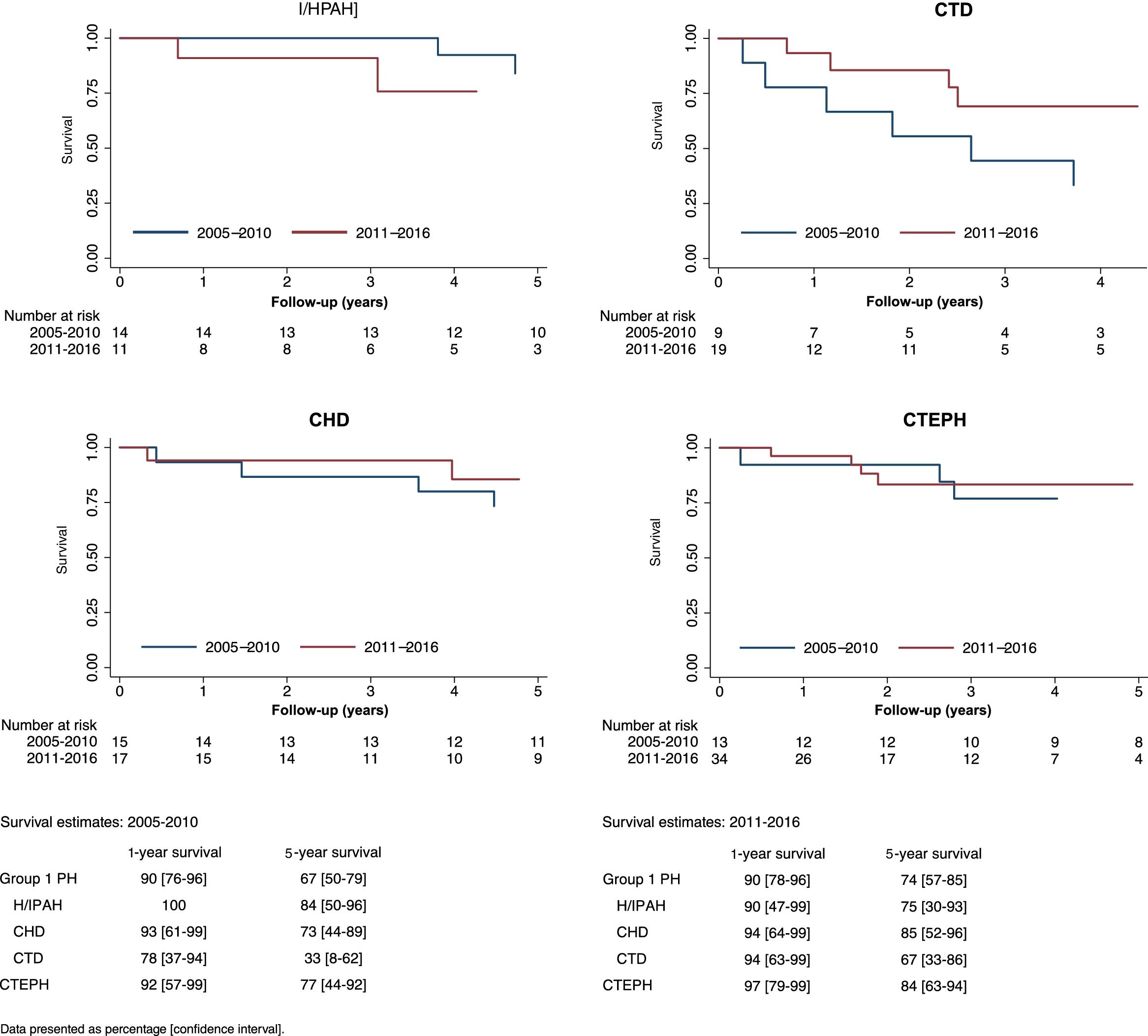
Survival curves of idiopathic/heritable pulmonary arterial hypertension, connective tissue disease-associated pulmonary arterial hypertension, congenital heart disease-associated pulmonary hypertension and chronic thromboembolic pulmonary hypertension patients by time periods of diagnosis (2005-2010 and 2011-2016). CHD: congenital heart disease; CTD: connective tissue disease; CTEPH: chronic thromboembolic pulmonary hypertension; I/HPAH: idiopathic/heritable pulmonary arterial hypertension; PH: pulmonary hypertension.
These long-term data of PH patients followed at a Portuguese referral center illustrate the following: (1) at diagnosis most of patients were in an advanced NYHA functional class; (2) the mean age at diagnosis of I/HPAH patients is increasing over time; (3) specific treatments and survival are comparable to other registries and cohort studies; and (4) PEA and lung transplantation are underused treatments despite being associated with better survival of CTEPH and PAH patients.
Most patients, 64% of group 1 PH and 57% of CTEPH, presented in NYHA functional class III-IV at diagnosis. Over time, a reduction was observed in the proportion of patients with advanced functional incapacity only in the CTEPH subgroup (92 vs. 44%, p=0.004). Observational data from registries in the UK, France and the USA that enrolled a broad spectrum of PAH etiologies consistently showed that patients diagnosed in NYHA class I and II had lower mortality than those in NYHA class III and IV.5,12,13 The earlier detection of CTEPH in our patients in the more recent study period might be explained by clinicians’ and patients’ increased awareness of this disease due to local and international campaigns.14–16 Despite our continued efforts, more has to be done to diagnose patients earlier, particularly in group 1 PH, such as establishing screening programs in high-risk populations and raising awareness of the disease. Currently, PH care in Portugal is organized into four regional treatment centers to which patients with suspected PH should be sent for assessment. Further development of this referral network might help the early diagnosis of PH.
Changes in the demographic landscape of PAH have been consistently described by several contemporary registries.17,18 Our data corroborate the higher mean age at diagnosis of PAH, a disease classically described as affecting young women. In addition, in our study age was independently associated with prognosis. This trend may reflect increased life expectancy, the widespread availability of screening tools like transthoracic echocardiography, and growing awareness regarding PAH in the medical community.19 Irrespective of the underlying cause of this demographic shift in PAH patients, this subset of older patients poses specific clinical challenges regarding their response to pulmonary vasodilators and their potential side effects, which need to be addressed in future studies.
In line with previous studies,5,20 the PH patients with the worst prognosis were those with CTD-PAH, who presented a five-year survival of 52%. Prognosis in this subset of patients showed improvement over time (Figure 2) despite the similar functional class at diagnosis in the two periods (NYHA III-IV class: 67 vs. 68%; p=0.93). We hypothesize that a more aggressive treatment approach can, at least in part, explain this better survival. In contrast, survival of patients with I/HPAH was unexpectedly worse in the more recent study period. Given the similar aggressiveness of vasodilator treatment in this subgroup over time, we speculate that the higher percentage of vasoreactive PAH patients (36% vs. 9%) can explain the worse survival. This difference may reflect survival bias, given the better prognosis of vasoreactive PAH. The overall survival rates of this subgroup (one-year and five-year survival rates of 95% and 80%, respectively) were similar to those of other registries,21 including one recently published by another Portuguese referral center.11
At the time of death, only 20% of PAH patients were under parenteral prostanoid therapy. The underuse of parenteral prostanoids in severe PAH has been reported by others.22–24 Physicians overlooking disease progression and underestimating its severity, organizational deficiencies in the management of this complex therapy, late referral, and patients’ refusal may all contribute to the underuse of prostanoids in PAH.25 Patient refusal, particularly by older patients, and social and cultural factors hindering self-management of continuous parenteral therapy were the main obstacles we found in our clinical practice. The lung transplantation numbers in our cohort reflect the constraints on this very effective treatment for PAH in Portugal, given the low volume of lung transplantations performed, particularly for PAH patients. This is a major issue that should be dealt with in the near future, by collaborating with national surgical centers or with foreign PH expert centers in the European Reference Network on respiratory diseases (ERN-LUNG) recently approved by the European Commission.26
As in previous reports from Portugal,9,11 CHD-associated PAH represented a significant proportion of our cohort, which may be due to the limited access to corrective cardiac surgery in the past. Despite the lower age and better functional class at diagnosis, and a trend toward better survival over time, efforts should be made to enhance the prevention of PH development by early referral of these patients to the recently designated congenital heart disease expert centers network.27
Estimated one-year and five-year survival in our CTEPH patients was 95% and 81%, respectively. These outcomes are in line with those reported in other registries (one-year: 88-97%; five-year: 65-83%).9,28–31 PEA is the cornerstone treatment for CTEPH patients.2 It is potentially curative and is associated with low in-hospital mortality if performed in high-volume centers.32 Despite robust evidence of its benefits, underuse of PEA has been reported in previous registries. Escribano-Subías et al.28 showed that this underuse is particularly marked in centers without PH expertise, in which only 5% of CTEPH patients underwent PEA, compared to 48% of those treated in expert centers. Almost 40% of our CTEPH patients underwent PEA or angioplasty after referral to a European high-volume surgical center. After adjusting for age, PEA was no longer associated with better survival, suggesting that some of the observed survival differences between operated and non-operated subgroups are accounted for by patient characteristics. The low statistical power, and consequent wide confidence intervals, should also be taken into consideration when analyzing these results. The main reason for not undergoing PEA was patient refusal to undergo the procedure in a foreign center, despite the existence of a national program that covers all expenses of cross-country care in this field. The majority of non-operated patients were treated with pulmonary vasodilators, initially with an endothelin receptor antagonist and later with a soluble guanylate cyclase stimulator, recently approved for the treatment of non-operable patients and those with persistent PH after PEA.2
Our study has several limitations that should be considered. The single-center nature of our data limits the generalizability of our conclusions, as etiologies and access to PH treatments may vary significantly across different centers. The small number of patients in some of the subgroups limits wider inferences. Despite the strong signs of improvements in survival, our analysis is underpowered to detect statistically significant time trends in the survival of patients diagnosed at different periods.
ConclusionsOur data consistently indicate a trend toward improved survival over time of PH patients treated at a Portuguese referral PH center, except for I/HPAH. Efforts to achieve an early diagnosis, to increase use of parenteral prostanoids and to promote the delivery of surgical treatments to PH patients are needed to further improve their prognosis.
Conflicts of interestThe authors have no conflicts of interest to declare.
None.
