





A 51-year-old woman presented with a one-year history of progressive dyspnea, WHO functional class III-IV and exercise-related syncope. Transthoracic echocardiography and computed tomography pulmonary angiography were performed, leading to a diagnosis of pulmonary arterial hypertension. She was referred to our pulmonary hypertension unit, where a complete study was performed, including ventilation/perfusion scan, which was consistent with chronic thromboembolic pulmonary hypertension. Risk factors for this condition were excluded and therapeutic options were evaluated. Imaging studies showed distal pulmonary disease so pulmonary endarterectomy was rejected. Further therapeutic options were evaluated and the patient was subsequently enrolled in an open-label uncontrolled trial with riociguat. After one year of treatment, significant improvement in functional class, 6-minute walk test and NT-proBNP were seen, without significant secondary effects.
Uma mulher de 51 anos de idade apresentou-se com história de um ano de dispneia progressiva com classe funcional III-IV e síncope relacionada com exercício. A ecocardiografia transtorácica e a angiotomografia pulmonar computadorizada foram realizadas, tendo sido diagnosticada hipertensão arterial pulmonar. A doente foi encaminhada para a Unidade de Hipertensão Pulmonar, onde foi repetido o estudo completo, incluindo verificação de ventilação/perfusão, compatível com hipertensão pulmonar tromboembólica crónica. Os fatores de risco para essa situação foram excluídos e foram avaliadas as opções terapêuticas. Estudos de imagem mostraram doença pulmonar distal pelo que a endarterectomia pulmonar foi rejeitada. Foram avaliadas novas opções terapêuticas e a doente foi posteriormente inscrita num estudo aberto, não controlado, com ensaio com riociguat. Após um ano de tratamento, uma melhoria significativa na classe funcional, um teste de caminhada de 6 minutos e NT-proBNP foram alcançados sem efeitos secundários significativos.
Chronic thromboembolic pulmonary hypertension (CTEPH) is an uncommon disease characterized by obstruction of the pulmonary vasculature by residual organized thrombi, leading to increased pulmonary vascular resistance, progressive pulmonary hypertension (PH) and, in its final stages, right ventricular failure. A ventilation/perfusion (V/Q) scan is mandatory for the screening of this condition. Long-term prognosis of patients with CTEPH is poor unless they receive treatment early. Pulmonary endarterectomy (PEA) is the treatment of choice for CTEPH and is the only potentially curative intervention, with low perioperative mortality in experienced centers. Previously, treatment options for patients ineligible for pulmonary endarterectomy were limited to off-label treatment with pulmonary vasodilators, balloon pulmonary angioplasty, and lung transplantation. However, riociguat, a novel oral soluble guanylate-cyclase stimulator, was recently shown to be an effective treatment option for patients with inoperable CTEPH, with improvements in prognostic variables such as the 6-minute walk test (6MWT), World Health Organization (WHO) functional class and N-terminal pro-brain natriuretic peptide (NT-proBNP). We present a case of inoperable CTEPH treated with riociguat.
Case reportA 51-year-old woman presented with a one-year history of progressive dyspnea and exercise-related syncope. The patient had no drug allergies or family history of note, but had a personal history of bipolar affective disorder (long-term treatment regimen consisting of oral lamotrigine 100 mg once daily and risperidone 6 mg once daily) and was a former smoker; she reported no other cardiovascular risk factors, use of anorectic drugs or consumption of toxic oils.
Two-dimensional transthoracic echocardiography showed dilated right heart chambers and signs of elevated pulmonary pressures, with no evidence of atrial septal defect and normal left ventricular systolic, diastolic and valve function. Computed tomography pulmonary angiography (CTPA) confirmed the presence of right heart chamber dilatation (right ventricular end-diastolic diameter of 51 mm) and signs of PH (pulmonary trunk diameter greater than that of the ascending aorta) with no visible pulmonary artery defects. As a result of these findings, the patient was diagnosed with pulmonary arterial hypertension (PAH) and was referred to the specialist PH unit at our center.
At admission to the PH unit the patient was in a wheelchair and reported WHO functional class III–IV dyspnea and exercise related-syncope for one year. Physical examination showed normal systolic blood pressure, heart rate and central venous pressure, with basal oxygen saturation of 85%. Auscultation of the heart revealed a loud second cardiac sound with an accompanying audible pulmonary valve regurgitation murmur. No physical signs of heart failure were found and abdominal examination was unremarkable. The electrocardiogram showed normal sinus rhythm, right atrial enlargement and right ventricular hypertrophy with secondary repolarization disturbances.
Subsequent investigations included a new transthoracic echocardiogram, pulmonary function tests (PFTs), arterial blood gases, full blood count (FBC), 6MWT and a V/Q scan. The transthoracic echocardiogram showed dilatation of the right heart chambers (right atrial area of 22 cm2 and right ventricular end-diastolic diameter of 44 mm), right ventricular wall hypertrophy (10 mm), right ventricular dysfunction (right myocardial performance index: 0.36; peak tissue Doppler velocity: 8 cm/s; and tricuspid annular plane systolic excursion [TAPSE]: 15 mm), diastolic dysfunction, normal systolic function with no mitral or aortic valve abnormalities, estimated pulmonary artery systolic pressure (PASP) of 115 mmHg and significantly increased pulmonary vascular resistance (pulmonary acceleration time of 49 ms). PFTs were carried out with the following results: forced vital capacity (FVC) 97%; forced expiratory volume in 1 s (FEV1) 100%; FEV1/FVC 68%; diffusing capacity of the lung for carbon monoxide (DLCO) 84%; DLCO/alveolar volume 93%; total lung capacity 109%; and residual volume 140%. Arterial blood gas results were: pH 7.41; PaO2 48 mmHg; PaCO2 35 mmHg; and SpO2 83%. FBC was normal. Serum NT-proBNP was significantly raised (1032 pg/ml). The patient was able to walk a distance of 180 m in the 6MWT without experiencing hypotension or desaturation.
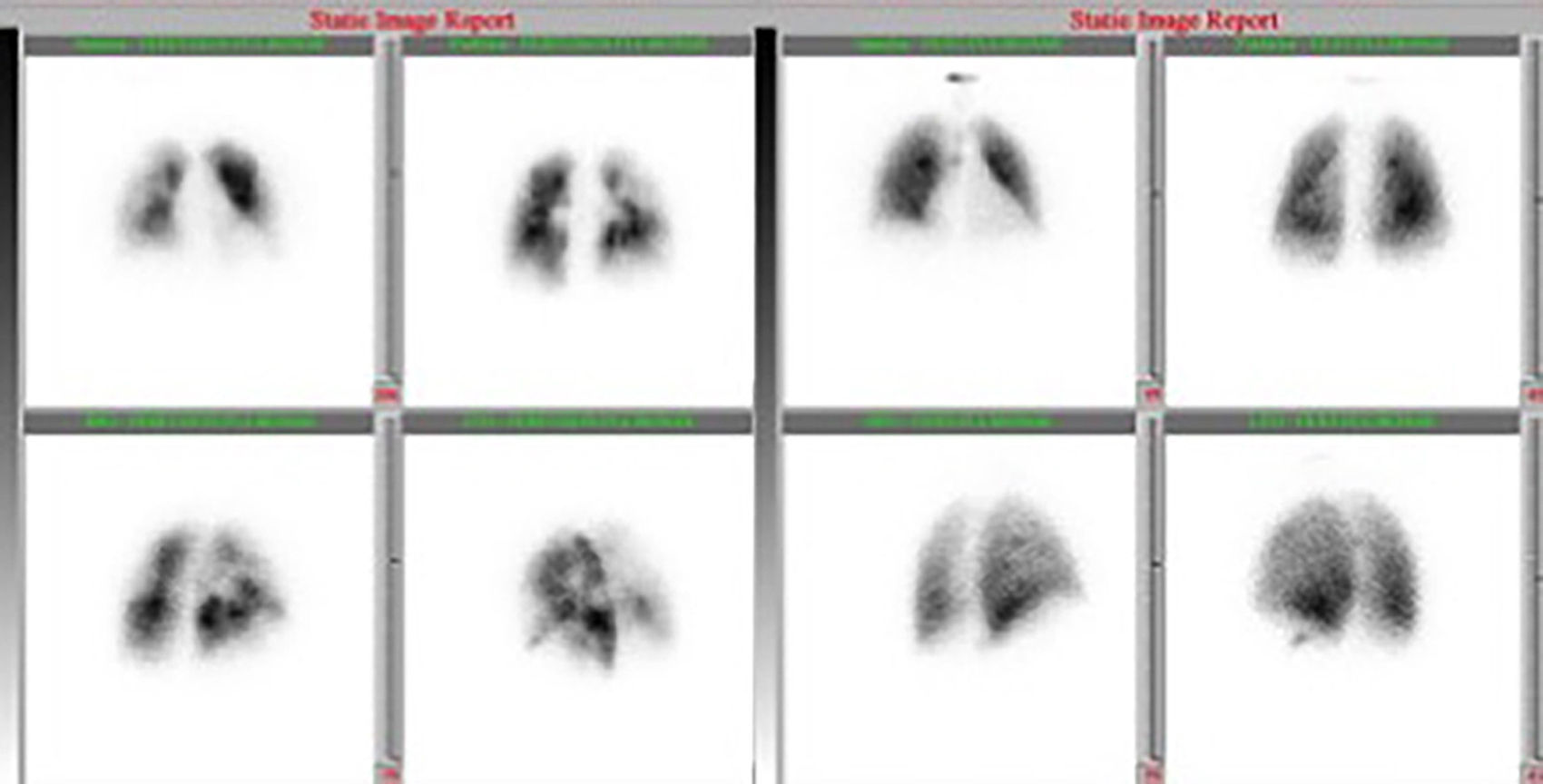
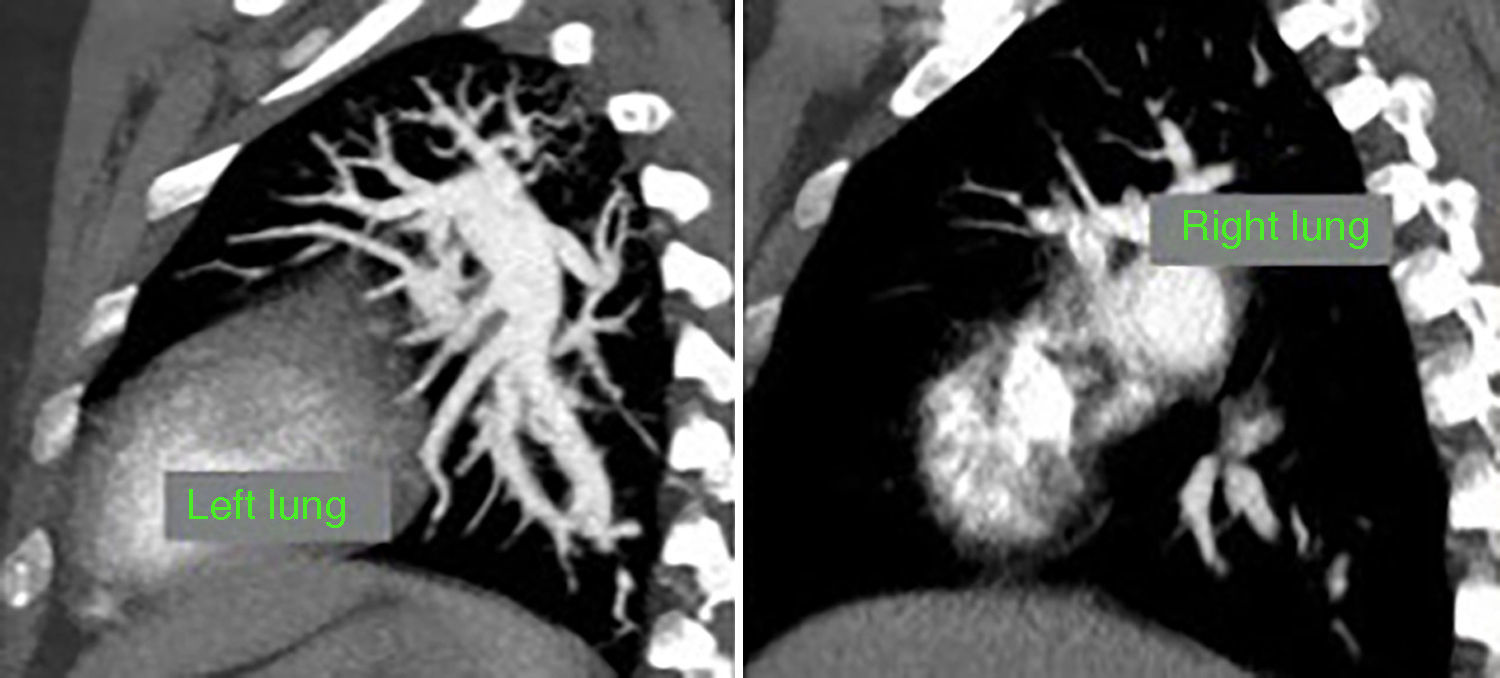
At this point, a new V/Q scan was performed in order to rule out CTEPH, despite the fact that previous CTPA showed no evidence of pulmonary artery defects. The V/Q scan showed multiple perfusion defects in both lungs, together with normal ventilation (Figure 1). Given the high sensitivity of V/Q scanning for detecting CTEPH, this positive result cast doubt on the original diagnosis. The next step was to perform a new CTPA, which now showed involvement of the left distal segmental arteries and the distal segmental branches of the right superior, mid and inferior lobar arteries (Figure 2).
Following a review of the results, the diagnosis was reconsidered and changed from PAH to CTEPH, confirmed by right heart catheterization and pulmonary angiography. Pulmonary angiography demonstrated the presence of very distal thrombotic occlusions and findings on right heart catheterization were compatible with CTEPH (mean right atrial pressure: 9 mmHg; right ventricular pressure: 135/19 mmHg; pulmonary artery pressure: 135/37 mmHg; mean pulmonary artery pressure: 72 mmHg; pulmonary capillary wedge pressure: 8 mmHg; cardiac output: 3.97 l/min; cardiac index: 2.64 l/min/m2; pulmonary vascular resistance: 3 Wood units).
The case was discussed with the specialist CTEPH team and the patient was considered unsuitable for PEA due to very distal involvement (University of California-San Diego surgical classification type III). Due to the lack of efficacy of classic vasodilators (endothelin receptor antagonists, phosphodiesterase inhibitors, and prostacyclins) in CTEPH, new therapeutic options were evaluated and the patient was subsequently enrolled in an open-label uncontrolled early access study for treatment with riociguat (an oral soluble guanylate cyclase stimulator).
The patient was started on a dose of 1 mg riociguat tid. The dose was increased by 0.5 mg tid every two weeks and any adverse events were reported. The maximum dose of riociguat (2.5 mg tid) was reached at visit 4 (week 6), in accordance with the study protocol. At this time the patient was in functional class III. At week 12, the patient reported that she was able to walk 200 m without effort, but that she had experienced an episode of exercise-related presyncope.
After three months on the maximum 2.5 mg tid dose of riociguat, the patient was able to attend her hospital check-up without wheelchair assistance or any other aids. At this visit, the patient had a 6MWT of 255 m and a Borg dyspnea index of 7, and was in functional class II. Between check-ups, she had not experienced new exercise-related syncope episodes, and no riociguat-related adverse events were reported. The patient was assessed again nine months after beginning riociguat treatment, at which time she walked 300 m during her 6MWT and had a Borg dyspnea score of 2.
At the last visit, one year after beginning riociguat treatment, the patient was in functional class II, and was able to walk 345 m during the 6MWT. She had suffered a new syncope episode in the context of severe anemia (hemoglobin 8.1 g/dl) due to gynecological bleeding, needing blood transfusion. Serum NT-proBNP was 367.3 pg/ml (Figure 3) and she showed no clinical signs of heart failure. Repeat transthoracic echocardiography was performed, and showed right chamber dilatation (right atrial area 22 cm2 and right ventricular end-diastolic diameter 40 mm), right ventricular systolic dysfunction (right myocardial performance index: 0.52, peak tissue velocity: 9 cm/s and TAPSE 17 mm) and PASP of 82 mmHg.
Discussion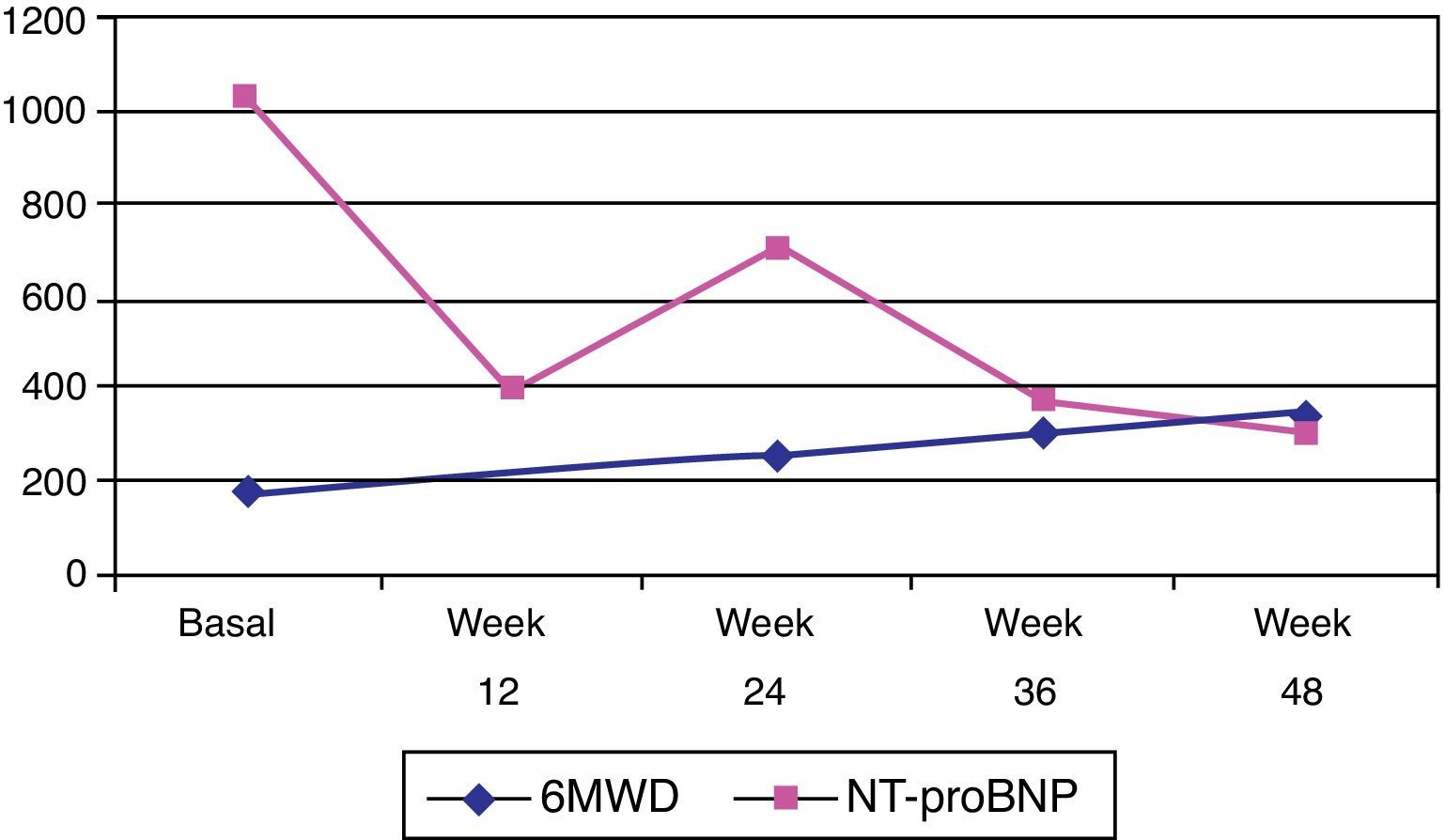
CTEPH is a disease caused by chronic obstruction of the pulmonary vasculature, which can occur in both the proximal and distal pulmonary arteries. CTEPH represents Group 4 in the recent WHO clinical classification of PH.1 CTEPH is defined as PH confirmed by right heart catheterization (mean pulmonary artery pressure ≥25 mmHg and pulmonary artery wedge pressure ≤15 mmHg), with the presence of chronic thrombi in the elastic pulmonary arteries after at least three months of anticoagulation therapy. Although the incidence of CTEPH is relatively low, it remains one of the most common subsets of precapillary PH.2
A number of risk factors have been identified for CTEPH, including previous splenectomy, hematological disorders (antithrombin deficiency, protein C and S deficiency, factor V Leiden, plasminogen deficiency, anticardiolipin and antiphospholipid antibodies, and lupus anticoagulant), ventriculoatrial shunts for treatment of hydrocephalus, indwelling catheters, cancer, treatment of hypothyroidism, and chronic inflammatory disorders. The clinical signs and symptoms of CTEPH are nonspecific and the diagnosis is therefore often delayed (with a median of 14 months to diagnosis from the onset of symptoms).3 V/Q scanning is the technique of choice when screening for CTEPH, due to its high sensitivity (>96%). This means that a normal V/Q scan essentially rules out CTEPH.4 Other useful diagnostic tools include CTPA and pulmonary arteriography, although they have lower sensitivity.
To date, the only potentially curative treatment for CTEPH is PEA, a surgical intervention with perioperative mortality rates of between <2% and 5% in experienced centers.3 In patients with inoperable CTEPH, balloon pulmonary angioplasty has been the subject of some research interest as a potential therapeutic option5; however, this technique requires further evaluation and has yet to become established.6 Bilateral lung or heart/lung transplantation, while potentially curative, is limited to young patients with no other treatment options. Until recently, the only available medical therapies for patients with inoperable CTEPH were oxygen therapy and lifelong oral anticoagulation. Pulmonary vasodilators, such as endothelin receptor antagonists, phosphodiesterase inhibitors, and prostacyclins, have not shown significant efficacy in patients with CTEPH in randomized trials.7,8 There is, however, a novel and promising pharmacological treatment, the oral soluble guanylate cyclase stimulator riociguat. In the recent phase III CHEST-1 trial, riociguat improved 6MWT, WHO functional class, NT-proBNP levels, and hemodynamic parameters in patients with CTEPH, with a good safety profile.9 Riociguat is the first drug approved by the European Medicines Agency and the US Food and Drug Administration for the treatment of CTEPH.
In this case report we show the clinical outcomes and safety profile of this new therapeutic agent in non-operable CTEPH cases.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that the procedures followed were in accordance with the regulations of the relevant clinical research ethics committee, with those of the Code of Ethics of the World Medical Association (Declaration of Helsinki).
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
Author contributionsProf. Gomez-Sanchez, Dr. Ortiz-Bautista, Dr. Navas-Tejedor, Dr. Morán-Fernández and Nuria Ochoa-Parra wrote the case report and prepared the manuscript.
Conflicts of interestProfessor Gómez-Sánchez has received speaker fees, honoraria, consultancy, advisory board fees, investigator, committee member, etc. from Bayer, Actelion, Pfizer, GSK and Ferrer Pharma.
Editorial assistance to carry out an English language check and copy edit of the final text was provided by Adelphi Communications Ltd (Bollington, UK), supported by Bayer Pharma AG, Berlin, Germany.
