



Inflammatory myofibroblastic tumor is a neoplasm with uncertain behavior. We describe a case in a 66-year-old female who underwent resection of a left atrial tumor suspected to be a cardiac myxoma which was subsequently diagnosed as an inflammatory myofibroblastic tumor. After three years’ follow-up the patient underwent a second operation to remove tumoral occurrence in the right atrium, diagnosed as an intimal sarcoma. It cannot be confirmed whether the tumoral recurrence with a different diagnosis (intimal sarcoma) was a progression from the primary tumor or the metachronous appearance of a spontaneous sarcoma.
O tumor miofibroblástico inflamatório é uma neoplasia de comportamento incerto. Descrevemos o caso clínico de uma mulher de 66 anos submetida à ressecção de uma massa tumoral na aurícula esquerda com suspeita de mixoma cardíaco, mas diagnosticada como tumor miofibroblástico inflamatório. Após três anos de seguimento, a doente foi operada pela segunda vez para ressecar um novo tumor na aurícula direita, diagnosticado como sarcoma intimal. A recidiva tumoral com um diagnóstico diferente de sarcoma intimal não pôde ser confirmada como progressão do primeiro tumor ou como aparência metacrônica de um sarcoma espontâneo.
Primary cardiac tumors are classified as benign, malignant or of uncertain behavior. Myxoma is the most frequent cardiac tumor and belongs to the group of benign neoplasms. Inflammatory myofibroblastic tumor (IMT) is considered to be of uncertain behavior, tending to produce local infiltration, but not frequent metastasis propagation, although it can be locally aggressive. Differential diagnosis is challenging due to the variability of its histopathologic myxoid and inflammatory cellular findings that are similar to myxoma and sarcoma. IMT does not display the cytologic atypia and nuclear hyperchromasia which characterize sarcomas, although a few mitotic figures can be found.1,2
Case reportA 66-year-old female was admitted to the hospital suffering progressive dyspnea for several months. Echocardiography revealed an intracavitary left atrial tumor. The report described a homogenous non-mobile mass of approximately 4 cm×3 cm originating from the interatrial septum and occupying nearly the entire left atrial cavity. The thoraco-abdominal computed tomography (CT) scan (Figure 1A) confirmed the presence of a left atrial tumor extending to the right inferior pulmonary vein outlet. Also, a hypodense Image 7 mm in size was seen in hepatic segment VI suggesting possible focal steatosis. The patient had no relevant family history. Her personal history included advanced diabetic retinopathy associated with neovascular glaucoma, without other relevant medical history.
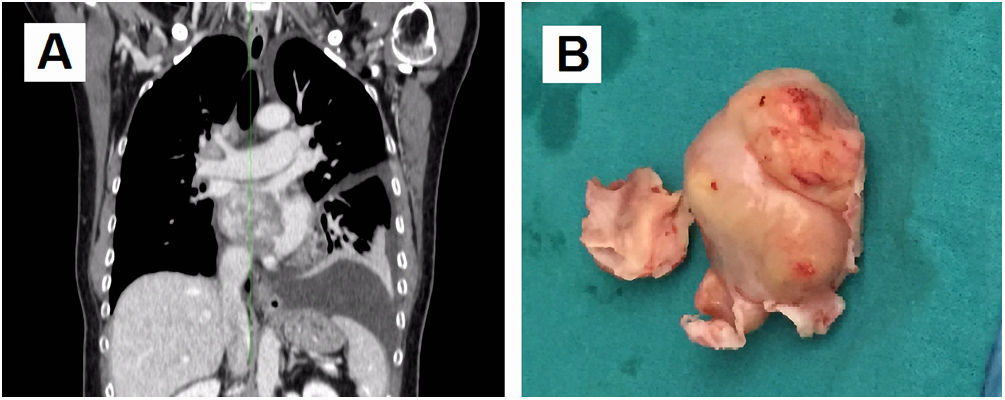
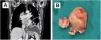
(A) Coronal view of thoraco-abdominal computed tomography scan before the first surgery. A left atrial tumor is observed extending into the right inferior pulmonary vein outlet; (B) mass resected during the first surgery, elastic in consistency with protuberant characteristics and mother-of-pearl surface color. Part of the interatrial septum can be observed at its base.
Surgery was performed (Figure 1B) establishing cardiopulmonary bypass with aortic and bicaval cannulation. The surgical approach was biatrial: the first approach was the left atrium via the interatrial groove, but direct right atrial access was required later for complete resection of the tumor and its base of implantation. The tumoral implantation was found in the left atrial face of the interatrial septum in the area corresponding to the foramen ovale. With a diameter estimated at 6 cm×5 cm, it occupied almost all the available space in the left atrium and expanded into the origin of both right pulmonary veins. The tumoral implantation zone extended inferiorly to the coronary venous sinus orifice. The mass displayed a mother-of-pearl appearance and elastic consistency with protuberant characteristics. A clear cleavage plane was identified by a fibrous sheet which allowed a dull dissection, separating the mass from the atrial wall, making the suspicion of myxoma controversial and less likely. Resection was completed including the base of implantation. Reconstruction of the interatrial septum with a pericardial patch was necessary.
The postoperative course was marked by the need for permanent pacemaker implantation due to complete atrioventricular block. Echocardiography demonstrated total absence of the atrial tumor and good biventricular function. The study was extended with positron emission tomography, which showed no signs of activity suggesting metastasis, including the hepatic lesion seen in the preoperative thoraco-abdominal CT scan. Anatomopathological study (Figure 2A) led to the diagnosis of an IMT with spindle cells expressing actin, calponin and CD68; no atypia or mitosis was reported. Tests for pS100, anaplastic lymphoma kinase (ALK), p53, immunoglobulin G4, Epstein-Barr virus and calretinin were negative. A few zones of spindle cells were associated with fibrohistiocytes and giant multinucleated cells, some of them with mitosis.
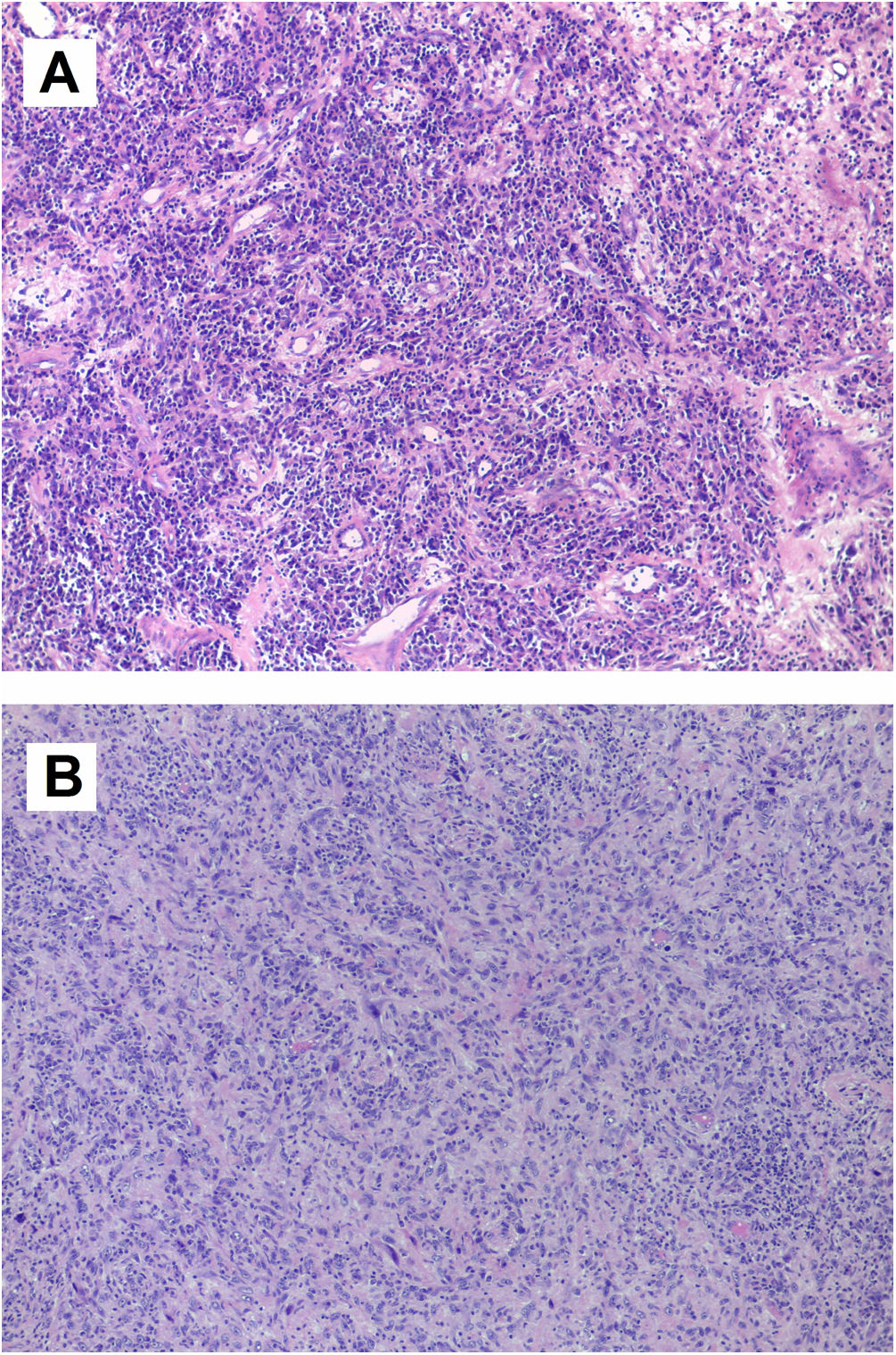
(A) Microscopic image of inflammatory myofibroblastic tumor. Tumor lesion of heterogeneous cellularity in a loose stroma, which varies from mucoid to dense with abundant collagen. It is vascularized by dilated vessels of variable caliber, some thrombosed. The predominant cells are lymphocytes and plasma cells, polyclonal inflammatory in nature, which mask the stromal cells. There is no atypia or mitosis. Other areas contain fibrohistiocytes and multinucleated giant cells, some undergoing mitosis; (B) microscopic image of intimal sarcoma. Proliferation of elongated spindle and stellate cells that grow interconnected in long, ill-defined bundles infiltrating the cardiac muscle. The cells show marked nuclear pleomorphism, without necrosis. There are three mitoses per 10 high-power fields.
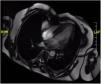
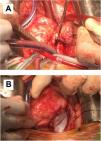
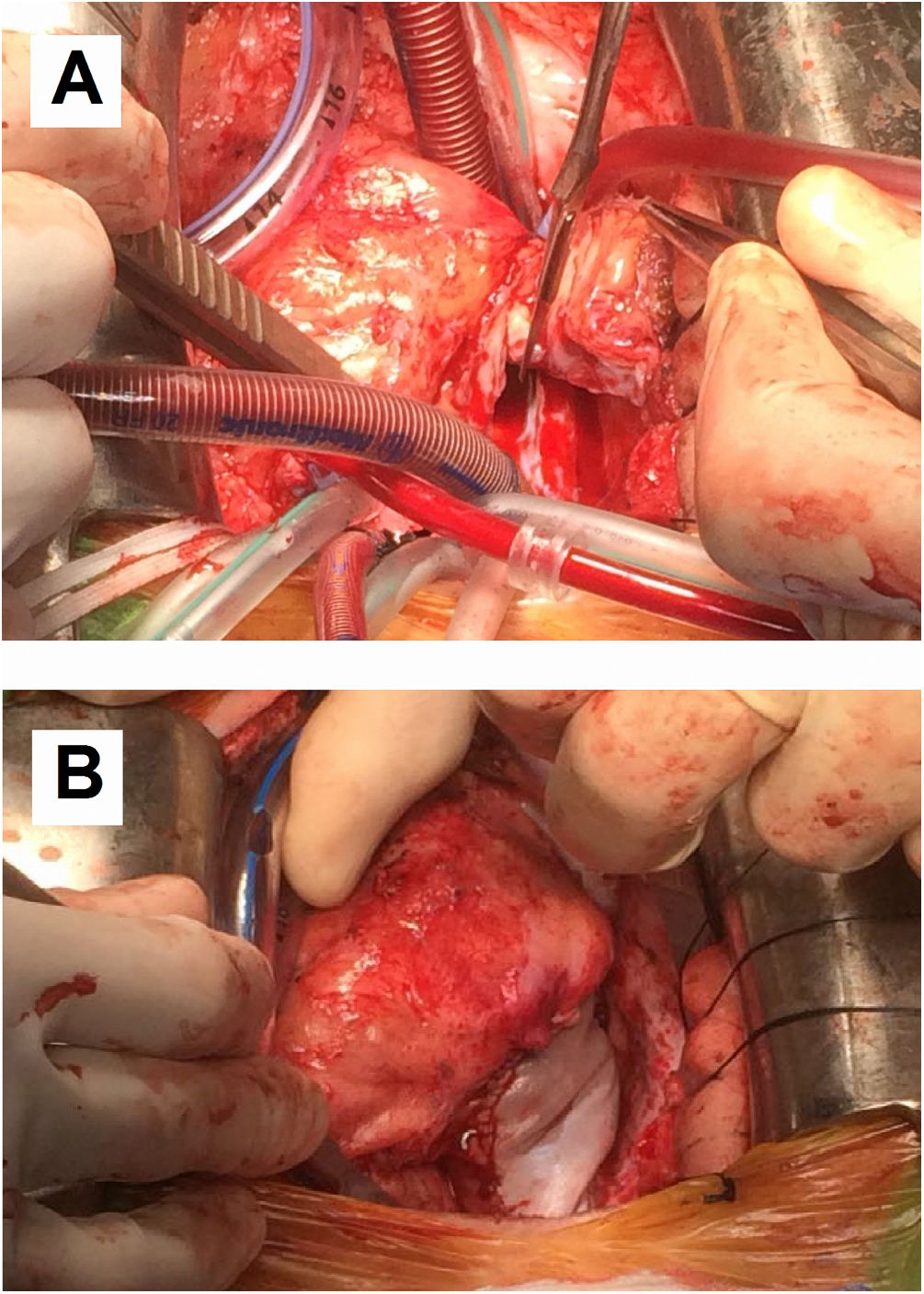
Although postoperative echocardiographic controls were normal, a CT scan in the third follow-up year revealed a contrast-enhanced mass occupying the right atrium measuring 17 mm×15 mm×34 mm with no infiltration to the surrounding structures. There were no other relevant findings. Magnetic resonance imaging was performed (Figure 3) to specify the mass's characteristics; it was isointense in T1 and presented edema and late contrast enhancement, suggesting malignant properties. Although the patient remained asymptomatic, after assessment by the heart tumor committee it was decided to operate due to the tumor's resectable characteristics. Resternotomy was performed (Figure 4), bicaval cardiopulmonary bypass was initiated and right atriotomy revealed a solid and homogeneous tumoral mass occupying the right atrial free wall extending from the superior to the inferior vena cava. Its surface and interior had a mother-of-pearl appearance and no necrosis or hemorrhagic areas were observed. Resection was complete although no clear boundaries were identified. Reconstruction of the right atrial free wall with a heterologous pericardial patch was performed.
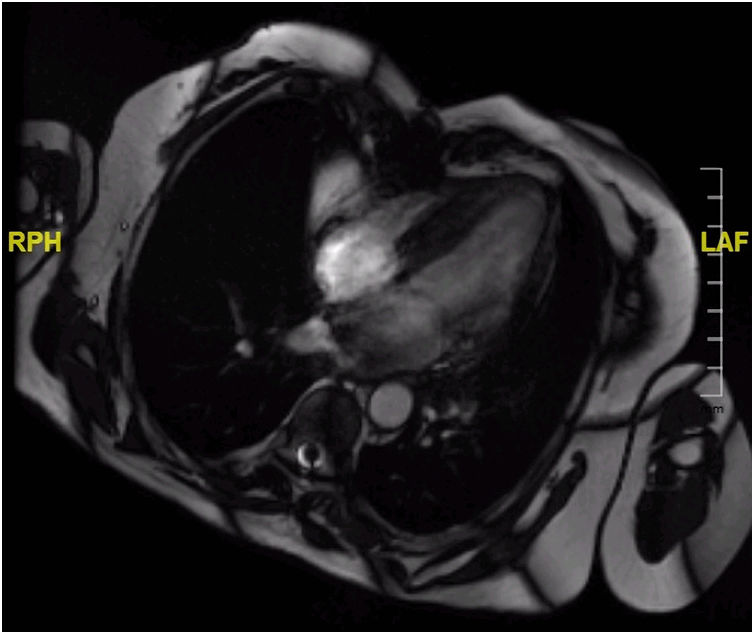
Postoperative echocardiography confirmed the absence of residual right atrial masses and normal ejection fraction in both ventricles. However, during the CT control scan three months after the second intervention, a new increase in right atrial free wall density was reported, suggesting tumoral recurrence, with suspected pericardial infiltration. Magnetic resonance confirmed this finding. Surgical biopsy (Figure 2B) revealed spindle and stellate cells infiltrating cardiac muscle with marked nuclear pleomorphism without necrosis and a rate of three mitoses per 10 high-power fields. Immunohistochemistry and fluorescence in situ hybridization study demonstrated MDM2 overexpression, compatible with a mesenchymal neoplasm suggestive of intimal sarcoma. Chemotherapy was not initiated due to the absence of changes in the immediate postoperative period on the CT scan, cardiac magnetic resonance and echocardiography performed during follow-up, thereby avoiding the side effects of chemotherapy that would have been administered with palliative intention. The last control was a cardiac magnetic resonance in the 10th postoperative month. The patient remained asymptomatic in cardiorespiratory terms, without dyspnea or congestive symptoms.
DiscussionPrimary cardiac tumors are rare, and almost 70% of them are benign. Myxoma, the most frequent heart tumor in adults, is normally found in the left atrium, which makes it the first diagnostic hypothesis when a mass is encountered there.1,3 IMT is considered to be underdiagnosed. It is characterized by a variety of myxoid and inflammatory findings with a mitosis rate of less than one per 10 high-power fields. ALK expression is not a specific marker but is one of its characteristics.1
IMTs appear more frequently in the lungs than in the heart and usually manifest during childhood or adolescence.4 The tumor's recurrence rate has not been thoroughly studied in the literature due to its low incidence.1 It is considered to be of uncertain behavior, tending to produce local infiltration, but not frequent metastasis propagation, although it can be locally aggressive.1,2 The natural history of IMT is benign and surgical resection is considered curative. Thus, surgical intervention is recommended if the mass is deemed to be susceptible to successful complete resection, given its potentially fatal outcome if prolapse or embolization occurs. Nevertheless, close follow-up is advised due to its unpredictable subsequent behavior.1,2,4
A negative finding of ALK expression in IMT is associated with aggressive behavior. Cases have been reported with a higher frequency of local recurrence and distant metastasis. Malignant transformation is possible in such situations, when these tumors are classified as sarcomas. In addition, they usually present a worse response to treatment.5–7
ConclusionWe present a case report of a 66-year-old female in whom surgical resection of a left atrial tumor was performed. Initially, a cardiac myxoma was suspected given its frequency and typical left atrial location. However, the macroscopic characteristics, microscopic findings and immunohistochemical study led to a diagnosis of IMT. After three years of follow-up the patient required a second intervention due to the appearance of a new mass in the right atrium, which was diagnosed as intimal sarcoma. It cannot be confirmed whether the tumoral recurrence with a different diagnosis (intimal sarcoma) was a progression and differentiation from the primary IMT or the metachronous appearance of a spontaneous sarcoma.
FundingNone declared.
Conflicts of interestThe authors have no conflicts of interest to declare.
