








Hypertrophic cardiomyopathy (HCM) is a genetically and phenotypically heterogeneous disease; there is still a large proportion of patients with no identified disease-causing mutation. Although the majority of mutations are found in the MYH7 and MYBPC3 genes, mutations in Z-disk-associated proteins have also been linked to HCM.
MethodsWe assessed a small family with HCM based on family history, physical examination, 12-lead ECG, echocardiogram and magnetic resonance imaging. After exclusion of mutations in eleven HCM disease genes, we performed direct sequencing of the TCAP gene encoding the Z-disk protein titin-cap (also known as telethonin).
ResultsWe present a novel TCAP mutation in a small family affected by HCM. The identified p.C57W mutation showed a very low population frequency, as well as high conservation across species. All of the bioinformatic prediction tools used considered this mutation to be damaging/deleterious. Family members were screened for this new mutation and a co-segregation pattern was detected. Both affected members of this family presented with late-onset HCM, moderate asymmetric left ventricular hypertrophy, atrial fibrillation and heart failure with preserved ejection fraction and low risk of sudden cardiac death.
ConclusionsWe present evidence supporting the classification of the TCAP p.C57W mutation, encoding the Z-disk protein titin-cap/telethonin as a new likely pathogenic variant of hypertrophic cardiomyopathy, with a specific phenotype in the family under analysis.
A miocardiopatia hipertrófica (HCM) é uma doença genética e fenotipicamente heterogénea, existindo ainda uma grande proporção de doentes sem uma mutação patogénica identificada. Embora a maioria das mutações seja encontrada nos genes MYH7 e MYBPC3, mutações em proteínas associadas ao disco Z também foram associadas à HCM.
MétodosAvaliámos uma pequena família com HCM com base na história familiar e exame objetivo, eletrocardiograma de 12 derivações, ecocardiograma e ressonância magnética. Após exclusão de mutações em 11 genes causadores de HCM, realizámos a sequenciação direta do gene TCAP que codifica a proteína titin-cap do disco-Z (também conhecida como teletonina).
ResultadosRelatamos aqui uma nova mutação do gene TCAP numa pequena família com HCM. A mutação identificada, p.C57W, mostrou uma frequência populacional muito baixa, bem como alta conservação entre as espécies. Todas as ferramentas de previsão de bioinformática usadas previram que essa mutação seria funcionalmente prejudicial. A presença desta nova mutação foi avaliada nos outros membros da família, tendo-se detetado um padrão de cossegregação. Ambos os membros da família afetados apresentaram HCM de início tardio, com hipertrofia ventricular esquerda assimétrica moderada, fibrilhação auricular e insuficiência cardíaca com fracão de ejeção preservada, com baixo risco de morte súbita cardíaca.
ConclusãoApresentamos evidência que suporta a classificação da mutação TCAP p.C57W, que codifica a proteína titin-cap do disco-Z (teletonina), como uma nova variante provavelmente patogénica da HCM, com um perfil fenotípico específico na família analisada.
Sarcomeric hypertrophic cardiomyopathy (HCM) is a monogenic disorder that is often familial.1,2 Transmission of the disease is autosomal dominant and it is caused classically by mutations in genes that encode cardiac sarcomere proteins.1,2 However, in patients with the HCM phenotype, the classic sequencing of sarcomere protein genes only identifies a disease-causing mutation in about 50-60% of cases.3 Intensive research is increasing the available genetic information available on this disease. Recently, formin homology 2 domain containing 3 (FHOD3) was proposed as a novel disease gene in HCM. It accounts for approximately 1% to 2% of all HCM cases.4 Mutations in Z-disk-associated proteins have also been linked to HCM5,6 and may partially explain some of the negative genetic tests for sarcomeric HCM. Several HCM-susceptibility genes, which encode Z-disk proteins, have been discovered, including actin alpha 2 (ACTN2),7 myozenin 2 (MYOZ2),8 muscle LIM protein (MLP),9 and telethonin (TCAP).10 Until the present moment, very few mutations in TCAP encoding titin-cap (also known as telethonin) causing cardiomyopathies have been identified (OMIM 604488).
We present the case of a family with HCM, in whom we excluded mutations in the eleven classic sarcomeric disease-causing genes. We further analyzed the TCAP gene, which led to the identification of the probable disease-causing mutation p.C57W, co-segregating with the family's phenotype.
MethodsPhenotypingThe two known affected members of the family were examined at an HCM tertiary referral center (Hospital da Luz – Lisbon, Portugal). Both individuals were followed up according to the guidelines,3,11 with ECG, echocardiogram, 24-h Holter monitoring, treadmill exercise test, and cardiac magnetic resonance (CMR), when available. These tests were performed at regular intervals and whenever there was a clinical indication. The HCM diagnosis was based on the established criteria.3,11–13 The local institutional review board approved the study and informed consent was obtained. The study protocol complies with the ethical guidelines of the 1975 Declaration of Helsinki.
Genetic analysisThe TCAP gene is located on chromosome 17q12 (assembly Genome Reference Consortium Human Build 38 patch release 13 (GRCh38.p13)), location NC_000017.11 (39665349…39666554). Genetic analysis of TCAP was performed as previously described.6,14 Genomic DNA was extracted from EDTA blood using the method described by Lahiri et al.15 Primer pairs were designed to amplify the two coding exons of TCAP with flanking intronic sequences based on the published sequence GenBank (at ncbi/HYPERLINK”http://nlm.nih.gov/genbank/”nlm.nih.gov/genbank/). While one primer pair was used for exon 1 (Tel1), we used three overlapping primer pairs for exon 2 to cover its large size of 393 bp (Tel2-4). All gene-specific sequences (18-20 bp) were preceded by 18 bp of M13 sequence (forward and reverse, respectively; M13F: TGTAAAACGACGGCCAGT and M13R: CAGGAAACAGCTATGACC) building 36 mer and 38 mer primers, respectively. The primer sequences were as follows:
Tel1F: CCCCATTAGTGAGTCTTGGC
Tel1R: GCTCAGTGAGGGTGCTCTGG
Tel2F: GGAGAGCAAAGGGGAACCAC
Tel2R: AGATGGGCAGCGGCAGTACC
Tel3F: CCTGGCTGATGATGCGGA
Tel3R: GGGACATGGAGCGGGACA
Tel4F: CTTCGTCGCTCCCTGTCC
Tel4R: CACCTCTTGCCCTCACCA
The final polymerase chain reaction mix (25 μl) contained about 20 ng of genomic DNA, 200 μM dNTP, 0.1 μM each primer, 0.625 U AmpliTaq Gold DNA polymerase and GeneAmp 10x PCR Buffer (100 mM Tris-HCl, pH 8.3, 500 mM KCl, 15 mM MgCl2, 0.01% (w/v) gelatin, Applied Biosystems, Darmstadt, Germany).14 The amplicons were analyzed by direct sequencing using ABI Big Dye Terminator chemistry and run on an ABI 3100 Avant device (Applied Biosystems, Darmstadt, Germany) as per the manufacturer's instructions. Detected variants in a sample were confirmed in at least two independent PCRs and sequencing runs. Sequencher software version 4.8 (Gene Codes, Ann Arbor, MI, USA) was used to facilitate data analysis and mutation identification followed by visual inspection of individual sequencing traces.6 Genetic variants were annotated according to the cDNA and protein reference sequence (Ensembl ID ENST00000309889.2 and UniProtKB/Swiss-Prot ID O15273).
The most common HCM-associated genes, the beta-myosin heavy chain (MYH7) and the myosin-binding protein C (MYBPC3) and other disease genes (TNNT2, TNNI3, TPM1, MYL2, MYL3, ACTC1, TNNC1, MYOZ2, and MLP) were systematically screened, as we and other colleagues have previously published.9,16–20 Disease-causing mutations in all eleven genes were excluded in the proband; other genes were not analyzed. The TCAP mutation was also excluded in 400 unrelated controls of Caucasian origin without hypertrophy and dilation and 400 subjects with dilated cardiomyopathy.14
Further analysis was performed by considering the frequency in population-based datasets, such as the Exome Aggregation Consortium (ExAC)21 and the Genome Aggregation Database (gnomAD).22 The latter consists of 141,456 human exomes and genomes (aligned against the GRCh37 reference). We also assessed the possible functional impact using seven different variant prediction algorithms, namely MutationTaster,23 M-CAP,24 PolyPhen2,25 SIFT,26 PROVEAN,27 Mutation Assessor28 and REVEL.29 These were not used in combination.
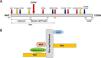
ResultsIdentification of the p.C57W mutationAfter exclusion of mutations in the eleven different HCM disease genes described above, we identified the novel heterozygous missense mutation c.171C>G (chr17:37822028C>G; dbSNP ID rs369447207) in exon 2 of the TCAP gene in one HCM patient, individual II-1, the proband of the identified family (Figure 1A and 1B). It leads to an exchange of tiny cysteine to large aromatic tryptophan at codon 57 (p.C57W), two amino acids with strong differences in their physical and chemical properties. The mutation affected both known TCAP transcripts (TCAP-201/ENST00000309889.2 and TCAP-202/ENST00000578283.1).
Mutation p.Cys57Trp in the TCAP gene. A: DNA sequencing electropherograms demonstrating heterozygosity for the detected mutation c.171C>G in individuals II-1 and II-2. The missense mutation is shown as two overlapping peaks (marked with an arrow). Individual II-3 exhibits a normal electropherogram (homozygous C). Codons are marked with gray blocks and the respective amino acid is shown below. B: Pedigree of the identified family. Squares represent males; circles females. Open symbols indicate unaffected individuals and solid symbols affected individuals; question marks, individuals with unknown status (without clinical data), and slanted bar, a deceased individual. The presence or absence of the mutation p.C57W is indicated by a plus and minus symbol, respectively. An arrow denotes the proband. C: Alignment of orthologs from eleven different species demonstrating high conservation in mammals (from chimp to dolphin) but no conservation in distantly related species such as fish. The mutated residue in the human sequence is underlined.
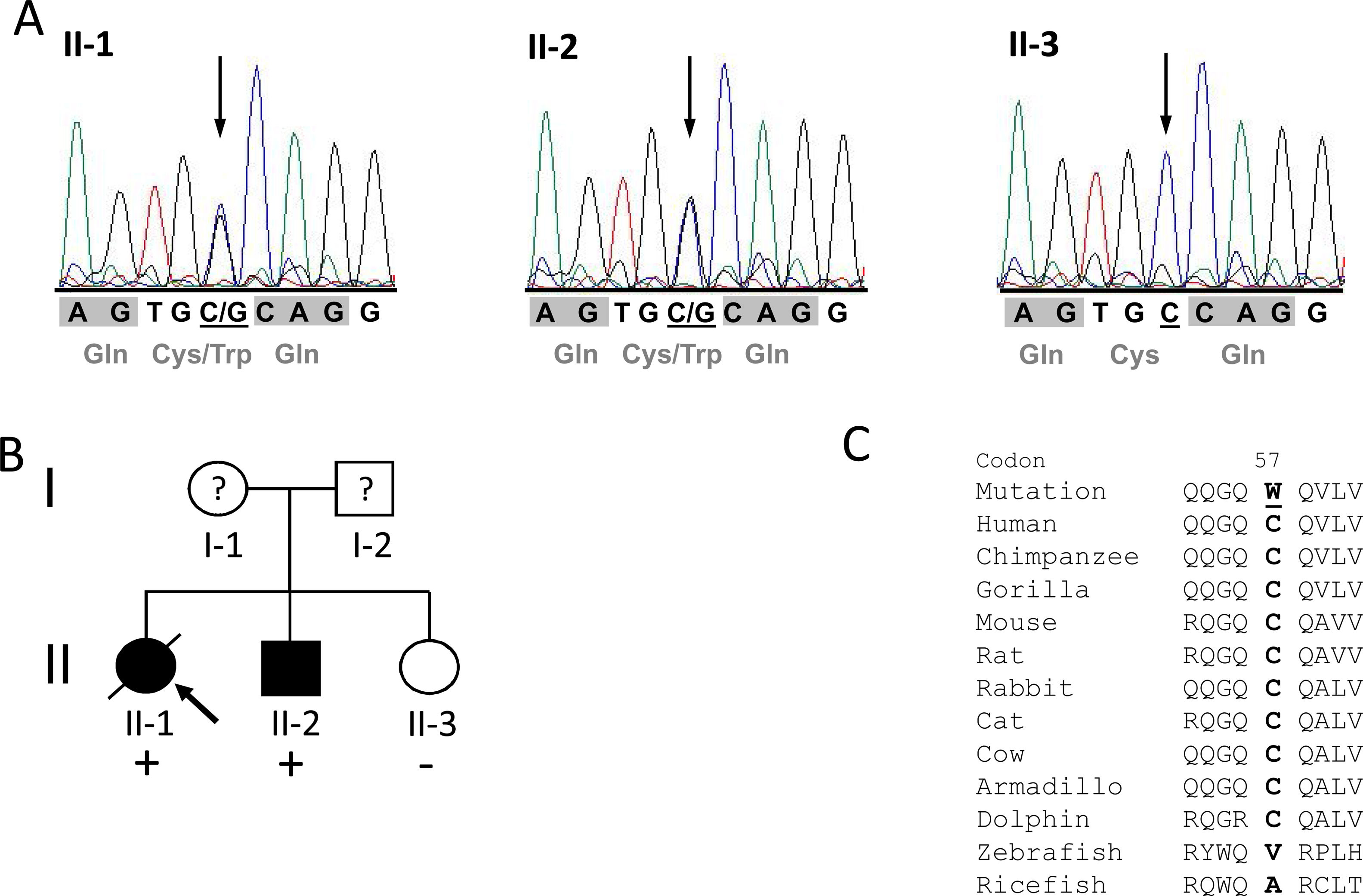
The mutation is located in overlapping domains of titin-cap/telethonin, which are involved in the binding of titin and muscle LIM protein (MLP), two important sarcomere proteins in the pathogenesis of cardiomyopathies (Figure 2A). The identified variant is very rare with a minor allele frequency (MAF) of 0.00003 (three alleles identified out of a total of 96 076 alleles analyzed) in the ExAC database as well as in gnomDB (four in 108 136; European population), confirming the assumption that pathogenic variants are very rarely detected in the general population. The mutation was also excluded in 800 control alleles we analyzed.
Distribution of mutations in the titin-cap protein and known interaction partners. A: The identified mutation p.C57W is located in the muscle LIM protein and titin interacting domain. All published mutations associated with cardiomyopathies are displayed. Red arrows indicate mutations in HCM patients and blue arrows mutations in dilated cardiomyopathy patients. Orange arrows denote three variations, which were initially described as mutations, but seem to be actually benign rather than disease-associated because of their frequency. The known interaction partners of titin-cap are shown below the bar representing the protein. B: Titin-cap interacts with a variety of different proteins in the Z-disk. In addition to the N-terminus of two titin molecules, it binds the potassium channel subunit minK, muscle LIM protein (MLP), and all three members of the calsarcin protein family (according to Frank et al.53). Please note we present only a selection of known interaction partners in this figure.
Alignment of different orthologs surrounding the affected codon 57 showed very high conservation through evolution in mammals (from chimp to dolphin), suggesting functional importance (Figure 1C). However, the residue shows no conservation in distantly related species such as fish. Further in silico analysis using seven bioinformatic prediction tools led to conclusive results concerning the identified mutation: all of them predicted the variant to be damaging/deleterious.
In addition to the proband, two further family members (individuals II-2 and II-3) were examined and analyzed. The phenotype co-segregates with the genotype: both mutation carriers were affected, while the family member without the mutation (II-3) had a normal phenotype (see pedigree in Figure 1B).
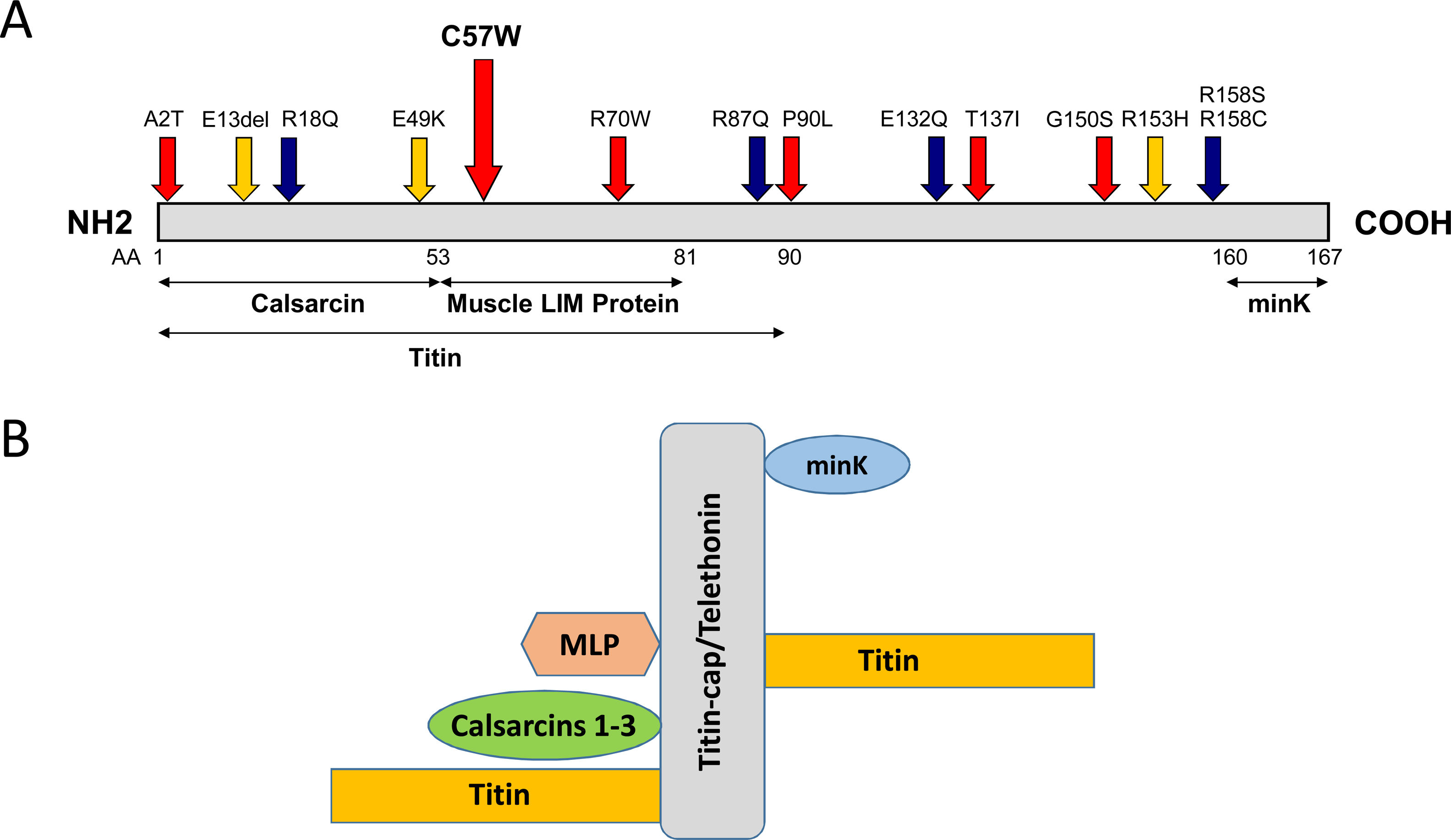
Clinical vignettesHypertrophic cardiomyopathy was first diagnosed in the female proband, individual II-1 (Figure 1B) at the age of 45 years when she developed paroxysmal atrial fibrillation (AF) after non-cardiac surgery. At that time, she was classified in New York Heart Association (NYHA) class II. Her physical examination revealed a systolic murmur at the left sternal border and aortic area that increased during orthostatism. The ECG showed sinus rhythm, left atrial dilatation, left axis deviation, pathological Q waves in aVF and DIII, V1 to V4, and SV2+RV6=35 mm, without ST-T changes. Transthoracic echocardiography showed asymmetrical hypertrophy of the anterior and inferior septum (neutral septal morphology) and lateral wall (maximal wall thickness 20 mm), without apical involvement (Figure 3). The left ventricle was non-dilated and left ventricular (LV) ejection fraction was preserved. The left atrium (LA) was dilated and there was evidence of elevated LV filling pressures (E/e′15+LA dilatation+tricuspid regurgitation velocity>2.8 m/s). The resting peak left ventricular outflow tract (LVOT) gradient was 61 mm Hg and mild to moderate systolic anterior motion (SAM) dependent mitral regurgitation was present. Systolic pulmonary artery pressure was estimated at 43 mmHg (38+5). Tissue Doppler imaging showed low s’ and e’ velocities at the mitral annulus. CMR was not performed. She began to take bisoprolol and oral anticoagulation (subsequently discontinued because of concomitant alcoholic liver disease) and remained clinically stable for 14 years. She died in 2003 at the age of 59, from a liver disease complication; she had no offspring.
Echocardiogram from the index patient II-1 (performed at the age of 54). Parasternal long axis view, two-dimensional and M-mode, showing asymmetric septal hypertrophy and mitral valve systolic anterior motion. Apical four chamber view and color wave Doppler show major left ventricular outflow tract obstruction. Please note that these 18-years-old echocardiographic images are suboptimal quality since they were obtained from still frames from thermal paper. Unfortunately, there are no better quality images available.
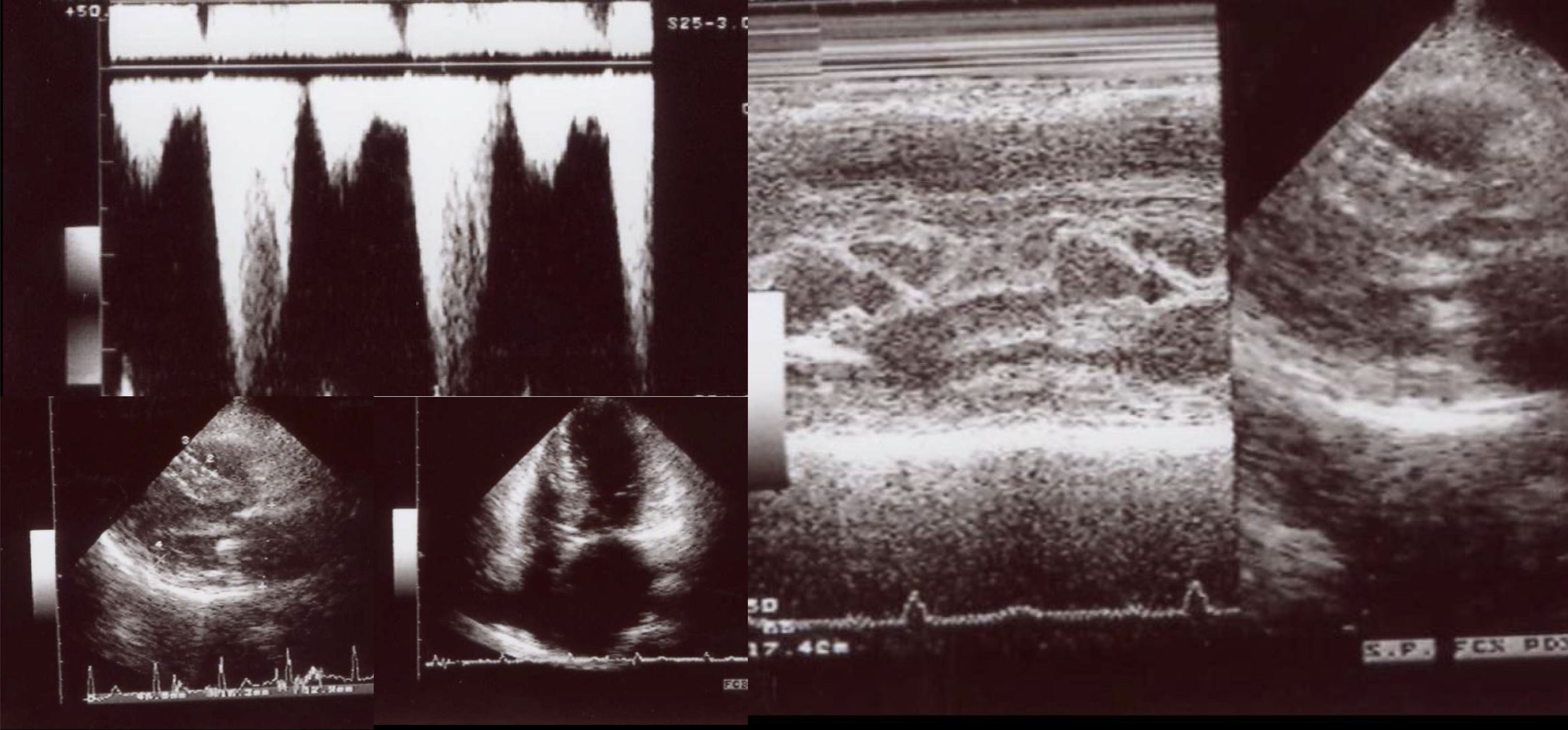
The affected brother (individual II-2), now aged 86, has an HCM phenotype, detected when he was 67 after genetic cascade screening. Although asymptomatic at diagnosis, he developed AF and has slow-progressing congestive right heart failure, now in NYHA III, with multiple hospitalizations in the past year. He reports no chest pain or syncope. His first ECG (Figure 4A) showed sinus rhythm, no LV hypertrophy criteria and marked ST-T changes in the precordial leads. His echocardiogram showed asymmetric hypertrophy, neutral septal morphology (21 mm maximal wall thickness) and no obstruction, neither at rest nor with bedside provocation maneuvers (Figure 5A). LV ejection fraction was preserved. When in sinus rhythm the E/e’ was 17. The right chambers were dilated and the right ventricle had reduced longitudinal function. He also had moderate pulmonary hypertension, 55 mmHg (45+10). His CMR revealed intramural late gadolinium enhancement in the hypertrophic segments, as well as in the insertion zone of the right ventricle in the lower interventricular septum (Figure 5B). Later, he developed AF, and right axis deviation (Figure 4B). There is no evidence of significant ventricular arrhythmias other than a single asymptomatic ventricular triplet in the 24-h ECG monitoring (Figure 4C). His European Society of Cardiology (ESC) HCM Risk-SCD is low (five-year risk <4%).3 Concomitant pulmonary embolism was excluded. He is currently taking rivaroxaban, bisoprolol, furosemide and spironolactone.
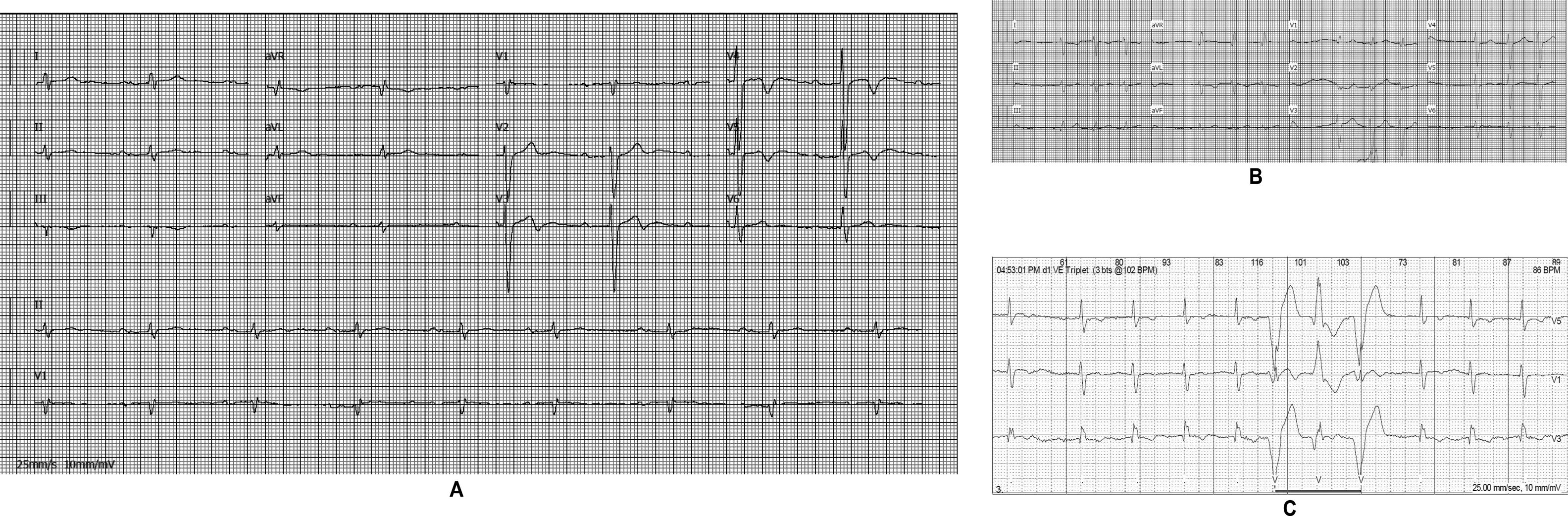
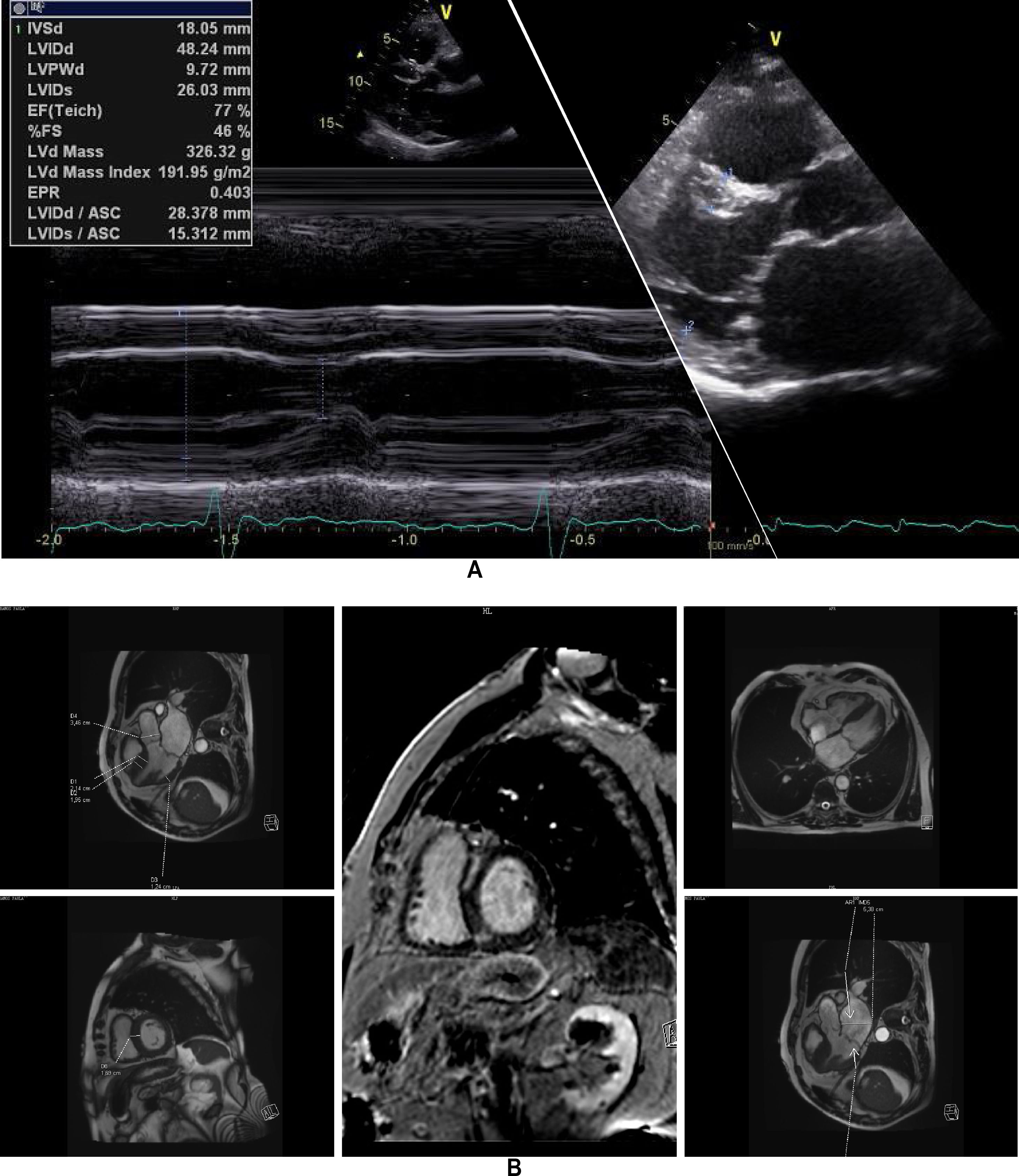
Echocardiogram and cardiac magnetic resonance imaging from patient II-2 (performed at the age of 84 years). A: Transthoracic echocardiogram, parasternal long axis view, 2-dimensional and M-mode, also showing asymmetric septal hypertrophy. B: These cardiac magnetic resonance images confirm the echocardiographic findings (asymmetric septal hypertrophy and mild mitral regurgitation) and provide tissue characterization data, with late gadolinium enhancement in the interventricular septum and in right ventricular insertion point in interventricular septum, typical findings of HCM.
The proband's sister (individual II-3) is 74 years old and exhibited no HCM phenotype. Her physical examination, ECG and echocardiography were normal. Her genetic test was negative for the p.C57W mutation in the TCAP gene.
DiscussionIn a small family of Portuguese origin, we identified the TCAP mutation, p.C57W. It demonstrated a very low population frequency and there is strong evidence for co-segregation with the HCM phenotype, as well as high conservation across species.
Considerations about the phenotypeSince there are only two patients in this family with the p.C57W mutation, it is impossible to draw definitive conclusions about clinical presentations and genotype-phenotype correlations. Additionally, as the index patient died prematurely of a non-cardiac condition, only a short follow-up of this patient is available.
Nonetheless, the two patients shared some common features: HCM was symptomatic in both and presented in adulthood, and was relatively late in onset. This corroborates data on the Portuguese population in the recently published Portuguese registry of HCM.30 The major common imaging features were: moderate asymmetric septal hypertrophy, dilated LA, increased LV filling pressures, symptomatic AF and heart failure with preserved ejection fraction. (Tables 1 and 2) There was no history of sudden cardiac death (SCD) or malignant ventricular arrhythmias. The ESC score-based SCD risk3 was low (Table 1), in contrast with data from two Japanese families with TCAP mutations, in whom three cases of SCD were found.10
Clinical and imaging data of the family members.
| Proband (II-1) | Affected brother (II-2) | Non-affected sister (II-3) | |
|---|---|---|---|
| ECG | LA dilatation, left axis deviation, pathological Q waves in avF and DIII, V1 to V4, LVH criteria, no ST-T changes | No LVH criteria, T wave inversion→right axis deviation | Normal |
| Echocardiogram | Septal+lateral wall hypertrophy, max 20 mmPreserved EFLVOTO: presentMild to moderate MR, SAM dependentLA dilation | Asymmetric septal hypertrophy, max 21 mmPreserved EFLVOTO: absentMV prolapse with mild MRLA dilation | No hypertrophy |
| Cardiac magnetic resonance | Not performed (died 2003) | LGE in areas of hypertrophy and in the insertion zone of the right ventricle in the lower interventricular septum | Not performed |
| Arrhythmias | Paroxysmal atrial fibrillation | Paroxysmal→Permanent atrial fibrillation | Not detected |
| Risk of SCD 5 years (ESC) | 2.21% | 2.61% | Not applicable |
EF: ejection fraction; LA: left atrium; LGE: late gadolinium enhancement; LVH: left ventricular hypertrophy; LVOTO: left ventricular outflow tract obstruction; MR: mitral regurgitation; MV: mitral valve; SAM: systolic anterior motion.
Detailed echocardiographic data of the family members.
| Proband (II-1) | Affected brother (II-2) | Non-affected sister (II-3) | |
|---|---|---|---|
| IVS (mm) | 20 | 21 | 9 |
| LVPWd (mm) | 12 | 8 | 8 |
| LVEDD (mm) | 43 | 48 | 47 |
| LVESD (mm) | 17 | 29 | 26 |
| LVOTO (mmHg) | 61 | 9 | 7 |
| LA-AP diameter M-mode (mm) | 43 | 55 | 34 |
IVS: interventricular septum; LA-AP: anterior-posterior dimension of the left atrium; LVEDD: left ventricular end diastolic diameter; LVESD: left ventricular end systolic diameter; LVOTO: left ventricular outflow tract obstruction; LVPWd: left ventricular posterior wall dimension.
Remarkably, two other mutations described in the literature are close to the one found in this study (Figure 2). The p.R70W mutation was described in a symptomatic HCM patient with massive hypertrophy and LVOT obstruction who subsequently underwent a myectomy.31 Interestingly, the patient was of Hispanic origin. Additionally, Hershberger et al. found the variant p.E49K in a patient with idiopathic dilated cardiomyopathy. The phenotype in this case was not described any further.32
Finally, Theis et al. studied 239 myofilament genotype negative HCM patients.33 In 13 of these subjects, they found a Z-disk mutation, and sigmoidal morphology was the predominant phenotype (85%). One of these 13 patients carried the p.R70W TCAP mutation, and this patient also had a sigmoid septum. These authors suggest that Z-disk HCM is associated preferentially with sigmoidal morphology. In the present study, however, we describe a family carrying a new TCAP mutation whose phenotype has neutral septal morphology.
TCAP mutationsAs shown by several studies, TCAP mutations identified in cardiomyopathy patients are distributed over the whole gene/protein10,31,33–38 (Figure 2A) and the differentiation between true pathogenic mutations and benign polymorphisms is challenging. One example is the in-frame deletion p.Glu13del initially described by Bos et al. as pathogenic,31 which we and many others have shown is benign because of its frequency.14,39 This was subsequently confirmed by a now publicly available resource, the ExAC database, showing a MAF of 0.00095 for this variant (115 found alleles out of 95 905 alleles analyzed in total). The same is true for p.R153H,10 which seems also too frequent to be a pathogenic variant (MAF 0.00017; 20/119 294 alleles). In contrast, the p.C57W mutation identified in this study is a very rare variant (MAF 0.00003) with similar frequency as the abovementioned p.R70W (MAF 0.00002). This is in line with the threshold MAF of 0.0001 for HCM suggested by Walsh et al.38
The mutation spectrum in the Portuguese HCM population seems similar to the published data. MYBPC3 and MYH7 are the most frequent disease genes, followed by cardiac tropin T (TNNT2) and/or cardiac tropin I (TNNI3), as demonstrated in studies by Cardim et al.,16 Brito et al.,40 Santos et al.,41 and Mendes de Almeida et al.42 Santos et al. analyzed 80 HCM patients using a panel of 28 HCM-associated genes, including TCAP, but no mutation was identified in that gene.41 Mendes de Almeida et al. screened 16 HCM patients for TCAP and 25 other HCM-associated genes and also failed to detect a TCAP mutation.42 It is notable that when analyzing individuals of Spanish origin, the study by Mademont-Soler et al. included a large cohort of 303 HCM patients who were screened by next-generation sequencing in 20 HCM-associated genes.43 They only found one TCAP variant of unknown significance (p.R33W) in a patient who also carried a known pathogenic MYH7 mutation. It is of interest that the TCAP gene was also screened in Portuguese patients with dilated cardiomyopathy (n=107) and this also revealed only one variant of unknown significance (p.E105Q).44,45TCAP mutations are very rare which makes the few identified variants and affected families all the more interesting and particularly intriguing.
Titin-cap is also involved in genetic skeletal muscle disease, autosomal recessive limb girdle muscular dystrophy type 2G (LGMD2G), which manifests in proximal muscles of the hip and shoulder girdles, and may be associated with cardiac involvement.46,47 One particularly noteworthy observation concerning our study is that among patients with LGMD2G due to titin-cap deficiency, the most commonly reported TCAP mutation p.Gln53Ter has been identified in only one patient from Portugal48 and Brazilian patients.49–51 The latter may be related to the long history of Portuguese emigration to Brazil.
Potential effects of the mutationTitin filaments form an uninterrupted connective link along the myofibers and are crosslinked at the Z-disk by titin-cap/telethonin.52 This small protein forms an antiparallel sandwich complex with two N-terminal domains of titin (referred to as Z1/Z2) at the Z-disk edge, demonstrating extraordinary mechanical stability, gluing two titin molecules together (Figure 2B).53–55 Titin-cap has a unique elongated 3D structure with a central, five-stranded, antiparallel β-sheet that is extended by two exposed wing-shaped β-hairpin motifs (A-B and C-D).53 The p.Cys57Trp mutation is located in the wing-2 motif (C-D). There, cysteine forms hydrogen bonds with arginine at position 33 of titin-cap. The structural integrity of the Z-disk depends upon the tight network of the hydrogen bonds between domains Z1/Z2 and titin-cap. These bonds enable the complex to resist extremely high mechanical loads.51,56 If tiny sulfur-containing cysteine is exchanged with large aromatic tryptophan, this may lead to a weakening or even a disruption of the core β-sheet structure of the titin-cap protein, which may disturb the binding of titin domains Z1 and Z2. This could subsequently lead to changes in the elasticity and stress resistance of sarcomeres. A possible correlate at tissue level may be myocyte disarray, a pathological hallmark of HCM.57 Unfortunately, we were not able to analyze this directly in this patient.
The p.C57W mutation is located in the domain (residues 53-81) binding MLP (Figure 2A and 2B), an important component of the stretch sensor machinery of the cardiac Z-disk.34 It is noteworthy that we9 and other authors31 have linked the mutations in CSRP3 encoding MLP to HCM.
To elucidate the true functional effects of the mutation, mechanistic studies, including the in vitro expression of the mutation in cultured cardiomyocytes, as well as in vivo expression in animal models (mouse, zebrafish, and drosophila) may be promising. Novel technology, such as CRISPR/Cas9, could be used to engineer precise knock-in models much faster than before.58 Another very promising approach for investigating the molecular mechanism of individual mutations underlying HCM is patient-derived induced pluripotent stem cells, from which cardiomyocytes can be derived in vitro.59 This was recently demonstrated by Feng et al. who reported that abnormal calcium handling properties underlie familial HCM caused by an MYH7 mutation.60
Study limitationsThere may be some limitations to the present study. Although we have excluded mutations in 11 known HCM disease genes, we cannot rule out the presence of other pathogenic variants in other genes. Functional studies may have helped to clarify the relevance of p.C57W, but this was beyond the scope of this study, and further investigations to elucidate its functional impact are needed. Although the location of the mutation in an important domain involved in anchoring the proximal end of titin (considering the known 3D protein structure) makes a functional effect highly probable, this consideration is so far only speculation. Further robust linkage was hindered by the small size of the family. However, the phenotype co-segregated clearly with the genotype. Additionally, the exclusion of disease-causing genes in the patient, the identification of a variant in an established HCM gene, the confirmation of a very low frequency, the high conservation of the affected amino acid, and the clear and conclusive results from the prediction tools strongly support our classification of p.C57W as a likely disease-causing mutation.
ConclusionsA novel TCAP mutation in a small family with HCM was identified and evidence supporting the classification of the mutation as a likely pathogenic variant was presented. The genetic diversity that underlies HCM, a prototypic single-gene disorder, remains a complex issue.1 Although TCAP mutations seem to be a very rare cause of HCM, we have highlighted another interesting TCAP mutation, and hope to shed some light on this underrepresented orphan HCM disease gene.
HCM is a sarcomeric disease but the genetic test for the classical disease-causing genes is negative in more than 1/3 of the patients. In recent years, mutations in Z- disk proteins have been proposed as potential additional disease-causing genes, but scientific evidence of this hypothesis is still scarce.
What does this study add?After exclusion of mutations in eleven different HCM disease genes, we present data supporting the classification of the TCAP missense mutation p.C57W encoding the Z-disk protein titin-cap/telethonin as a new likely pathogenic variant for HCM, with a specific phenotype profile in the analyzed family.
The authors have no conflicts of interest to declare.
This work was supported by Charité research grants, contract number HCMGen 01-2004.
