










Sepsis-induced cardiomyopathy is the leading cause of death in sepsis and is characterized by reversible myocardial depression. However, the specific mechanisms responsible for myocardial injury in sepsis are not known. The present study used bioinformatic analysis to explore the possible mechanisms of sepsis-induced myocardial injury and the therapeutic potential of curcumin.
MethodsThe GSE125042 microarray gene expression matrix was obtained from the Gene Expression Omnibus database, which includes 10 septic cardiomyocyte samples from cecum ligation perforation constructs and 10 sham-operated groups cardiomyocyte samples. Background correction and matrix data normalization were performed using the robust multiarray average algorithm. Differentially expressed genes (DEGs) screening was performed using the Limma R package expression matrix, and whole gene analysis was performed using the weighted gene co-expression network analysis R package to construct gene networks and identify modules. Enrichment analysis and gene set enrichment analysis was performed on the genes to be selected. Construct cellular and animal models of myocardial injury in sepsis were assessed and the effects of curcumin on a rat or cardiac myocytes were observed.
ResultsA total of 2876 DEGs were screened based on the GSE125042 chip, of which 1424 genes were upregulated and 1452 genes were down regulated. WGCNA analysis of the whole genes was also performed and a total of 20 gene modules were generated. Among them, the selected TLR1 gene was present in the most strongly correlated Brown module. Enrichment analysis of the upregulated DEGs with the Brown module showed that they were significantly enriched in biological processes related to ribosomal protein complex generation, cellular components related to phagocytic vesicles and molecular functions related to Toll-like receptor binding, affecting cardiomyocyte survival as a target for molecular intervention in septic cardiomyopathy. Animal experiments showed that curcumin reduced inflammation levels, improved cardiac function and increased survival in rats with septic myocardial injury. Cellular experiments showed that curcumin increased the survival rate of lipopolysaccharide-treated cardiomyocytes and down regulated TLR1 expression and inhibited NF-κB phosphorylation in cells in a dose-dependent manner. Molecular docking analysis revealed that curcumin interacted with TLR1 by hydrogen bonding and could be stably bound to inhibit the biological function of TLR1.
ConclusionOur study shows that curcumin attenuates myocardial injury in sepsis by inhibiting TLR1 expression, which provides a molecular theoretical basis for clinical treatment.
A miocardiopatia induzida por sepsis é a principal causa de morte por sepsis e é caracterizada por uma depressão reversível do miocárdio. No entanto, os mecanismos através dos quais a sepsis induz lesão do miocárdio não são conhecidos. O presente estudo utilizou uma análise bioinformática para identificar os possíveis mecanismos de lesão do miocárdio induzida por sepsis e avaliar o potencial terapêutico da curcumina.
MétodosO microarray de expressão génica GSE125042 foi obtida na base de dados Gene Expression Omnibus (GEO), que inclui 10 amostras de cardiomiócitos de animais com sepsis, induzida por perfuração e ligação do ceco (CLP), e 10 amostras de cardiomiócitos de animais controlo (Sham). A correção e a normalização dos dados da matriz foram realizadas recorrendo ao algoritmo robusto de média multiarray (RMA). A identificação dos genes diferencialmente expressos (DEGs) foi realizada usando a matriz de expressão Limma R e a análise de genes inteiros foi realizada através da análise ponderada de rede de coexpressão de genes (WGCNA) R para construir redes de genes e identificar módulos. A análise de enriquecimento e a análise de enriquecimento do conjunto genético (GSEA) foram realizadas nos genes a selecionar. Foi avaliado o efeito da curcumina na lesão miocárdica, em modelos celulares e animais de lesão sepsis.
ResultadosForam avaliados 2876 DEGs com base no chip GSE125042, dos quais 1424 genes estavam sobrerregulados e 1452 genes estavam subregulados. Foi também realizada uma análise WGCNA de todos os genes e foram gerados 20 módulos de genes. Entre eles, o gene selecionado TLR1 estava presente no módulo de Brown mais fortemente correlacionado. A análise de enrriquecimento dos DEGs sobrerregulados com o módulo Brown mostrou que estes foram significativamente enriquecidos em processos biológicos relacionados com a geração do complexo proteico ribossómico (BP), componentes celulares relacionados com vesículas fagocitárias (CC) e funções moleculares relacionadas os Toll-likereceptors (MF), que podem constituir alvos moleculares envolvidos na sobrevivência dos cardiomiócitos na cardiomiopatia séptica. Experiências com animais mostraram que a curcumina reduziu os níveis de inflamação, melhorou a função cardíaca e aumentou a sobrevivência em ratos com lesão séptica do miocárdio. Experiências celulares mostraram que a curcumina aumentou a taxa de sobrevivência de cardiomiócitos tratados com LPS e a expressão de TLR1 desregulada e inibiu a fosforilação NF-κB nas células, de uma forma dependente da dose. A análise da acoplagem molecular revelou que a curcumina interagia com TLR1 através de ligação de hidrogénio, o que poderia inibir a função biológica de TLR1.
ConclusãoO nosso estudo demonstrou que a curcumina atenua a lesão do miocárdio por sepsis ao inibir a expressão do TLR1, constituindo uma base teórica molecular para novas abordagens terapêuticas.
Sepsis is defined as a life-threatening organ dysfunction caused by a dysregulated host response to infection, associated with high mortality and morbidity.1–3 During its course, patients experience decreased systemic vascular resistance, which leads to left ventricular dilatation, decreased ejection fraction and diastolic dysfunction in order to maintain cardiac output, resulting in typical septic shock hypotension. The sustained shock leads to further myocardial cell injury and the release of endogenous injury-related molecular patterns.3,4
Although it is generally believed that heart defects caused by sepsis are remediable, septic cardiomyopathy, a serious complication of sepsis, aggravates the severity of the disease and leads to poor patient prognosis.5,6 Studies have found that mortality among sepsis patients without cardiovascular injury is <20%, while the mortality rate of sepsis patients with cardiovascular dysfunction is as high as 70%.7,8 Sepsis has become a major challenge in day-to-day intensive care unit treatment. The current treatment dilemma is how to improve the clinical outcome of patients with severe sepsis. Unfortunately, the number of severe sepsis cases is still increasing at a rate of 1.5% per year and has created a huge burden for public health resources.9
Curcumin, found in turmeric and its roots, is a natural polyphenol with anti-inflammatory, anti-cancer and anti-oxidation effects. Curcumin has potential therapeutic effects in allergies, diabetes, cardiovascular and cancer related diseases,10 and can reduce oxidative stress, inflammation and related cell death pathways for heart protection.11 Some studies have shown that curcumin inhibits lipopolysaccharide (LPS)-induced overexpression of inflammatory mediators in vascular smooth muscle cells by inhibiting toll-like receptor 4 (TLR4) expression, MAPK phosphorylation, and nuclear translocation of NF-κB to reduce myocardial injury.12
Studies have shown that TLRs and associated intracellular signaling molecule-mediated responses are important factors for the survival of septic animal models.13 Among them, TLR1 is one of the main TLR members, and its enhanced activity can also activate downstream NF-κB signal and cause inflammation.14 In addition, the literature indicates that TLR1 gene polymorphism can contribute to organ failure and prognosis in sepsis patients.15 We hypothesized that curcumin may play a therapeutic role by down regulating TLR1 to mediate apoptosis of myocardial cells in septic myocardial injury. Consequently, we conducted bioinformatic analysis on the myocardial transcription spectrum of sepsis chips in the GEO database to elucidate the gene expression of septic cardiomyocytes, then we also performed biological function experiments to detect the effect of curcumin on LPS-induced cardiomyocytes, and explored the molecular mechanism of curcumin on TLR1 through molecular docking experiments.
Materials and methodsFramework flow chartThe framework flow chart is shown below:
Data sources and quality control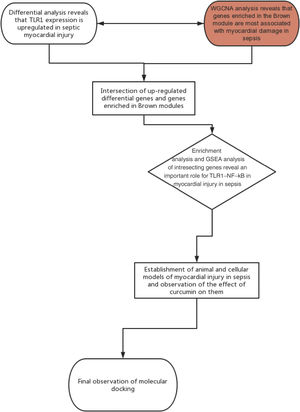
Chip GSE125042 expression matrix was obtained from the Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) database; these included 10 samples after cecum ligation perforation (CLP) induction (CLP group), and 10 samples as controls (Sham group).
Identification of differentially expressed genesBackground correction and matrix data normalization were performed using the robust multiarray average algorithm. Differential analysis of expression profiles between the Sham and CLP groups was performed using the Limma R package Wilcoxon test analysis, with ∣log2FC∣ >0.2630 and adj. p<0.05 as the screening condition.
Weighted gene co-expression network analysisCo-expression network analysis was performed using the weighted gene co-expression network analysis (WGCNA) R package to reveal the correlation of genes and determine positively related gene modules. The soft threshold power was set to 20; feature genes were then calculated, modules were clustered hierarchically and similar modules were merged.
UpSet plots moduleOnce the number in sets exceeds four, it is more difficult to produce a graphical output in the form of a Venn diagram. The Upset Venn diagram was plotted using UpSet R package, and the intersection between the up-down differentially expressed genes (DEGs) and WGCNA module genes in sepsis-induced cardiomyocyte was visualized using the Upset Venn diagram.
Enrichment analysisThe ClusterProfiler R package was used to enrich the up-regulated differentially expressed genes and modular genes, which was followed by gene ontology (GO) annotation and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis and the plot of GO clustering figure. The ggplot2 R package was employed to plot the GO secondary classification column and KEGG annotation statistical chart.
Gene set enrichment analysisThe up-regulated DEGs and modular genes were analyzed using the enrichplot R package, the enrichment scores (ES) were calculated to estimate the significative degree of ES and determine the biological significance of the gene set. Multiple hypothesis tests were therefore conducted to calculate the positive false discovery rate (FDR). FDR<0.25 and p<0.05 were used to analyze the false-positive rate of the value of expression.
Animal modelSixty healthy male Wistar rats were purchased from the Laboratory Animal Science Institute of Chinese Academy of Medical Sciences and placed in SPF environment (humidity 50±5%, temperature 20–22°C), of which 40 rats were subjected to LPS (Solarbio, 10 mg/kg). Intraperitoneal injection to replicate the rat model with septic cardiomyopathy, and the remaining 20 in sham operation group (Sham) were administered an intraperitoneal injection of the same amount of normal saline (Beijing Tiantan Biological Products Co., Ltd.). 20 rats with septic cardiomyopathy were injected intraperitoneally with curcumin (LPS+Cur) (Solarbio) at 100 mg/kg 1 hour before modeling. The remaining two groups (Sham, LPS) were injected with the same amount of normal saline. The rats were anesthetized for 12 hours, and then sacrificed with pentobarbital sodium (Shanghai Xinya Pharmaceutical Co., Ltd.) (200 mg/kg). The chest cavity was opened, 1 ml blood was taken from the heart, and myocardial tissue was frozen in liquid nitrogen for further determination. Thereafter, the survival of rats that could drink and eat freely were observed, and the survival rate of the three groups within seven days was calculated. All procedures were undertaken in accordance with the National Laboratory Animal Management Regulations and the National Laboratory Animal Management Implementation Rules.
Biochemical analysisAfter the blood was taken from the heart, it was left to stand at room temperature for 1 h, centrifuged at 3000 r/min for 5 min. The supernatant was taken, the levels of tumor necrosis factor-α (TNF-α), interleukin 6 (IL-6) and interleukin 10 (IL-10) in rat serum were detected using ELISA kit (abcam), the protein was expressed in pg/mg and the levels of brain natriuretic peptide (BNP), troponin I (cTnI), creatine kinase (CK) and creatine kinase MB (CK-MB) were detected using an automated biochemical analyzer.
Cell cultureMyocardial cells (H9C2) of the rat were obtained from Shanghai Zhong Qiao Xin Zhou Biotechnology Co. Ltd., all the samples were treated with 90% high glucose DMEM (Gbico), 10% FBS (Gbico) and 1% double antibody (Gbico), then incubated at 37°C, 95% air and 5% carbon dioxide. When the fusion rate reached 75%, the samples were treated with LPS (1 μg/ml) for 6 h to construct a model of LPS-induced myocardial injury.
CCK-8Twelve hours before adding LPS, each well was given different curcumin concentrations (0, 10, 30, 50, 100 μM), and six replicate wells were set in each group. CCK-8 kit (Solarbio) was used to detect the effect of curcumin on the proliferation activity of H9C2 cells after LPS treatment, then the cells were inoculated into 96-well plates (5*103 cells/well), 10 μl CCK-8 solution was added after 24 hours of culture. After incubation at room temperature for 4 h, the OD value at 450 nm was detected using microplate reader.
Annexin/PI double stainingUsing the same cell treatment as above, Annexin V-FITC an apoptosis detection kit (Procell) was used. Cells were seeded into six well plates, treated with LPS for 6 h, and washed twice with PBS. The cells then were resuspended in binding buffer and incubated with Annexin V-FITC/PI staining solution in the dark for 20 minutes at room temperature. The apoptosis rate of cardiomyocytes was analyzed using a C6 flow cytometer (BD).
Real time polymerase chain reactionThe total RNA in the cells was lysed using Trizol (Solarbio), the cDNA library was synthesized using the reverse transcription kit (Thermo), and then the target gene was amplified using the quantitative polymerase chain reaction (PCR) instrument Quantstudio7flex (Thermo). 2 μL cDNA, 10 μL 2×FastFire qPCR PreMix (Tiangen), a total of 0.6 μL upstream and downstream primers, and 6.8 μL RNase-Free Water were added to the system. An amplification reaction was carried out using the following condition: pre-denaturation at 95°C for 60 s, denaturation at 95°C for 5 s, annealing at 60°C for 15 s, 40 cycles. In this study, 2−ΔCT was used to calculate the relative level of expression of the target gene. The experiment was carried out three times and the average value was taken. All primer sequences were designed and synthesized by Shanghai GenePharma Co., Ltd. The primer sequences are shown in Table 1.
Western blotThe protein in the cells were extracted using protein kit (Solarbio), separated with 10% SDS-PAGE, transferred to PVDF film, then blocked with 5% skimmed milk at room temperature for 2 h, and washed three times (10 min every time) using TriTween buffer. Then the samples were added to the primary antibody (abcam) diluted by dilution buffer, incubated at 4°C overnight, washed three times with tris-buffered solution, and added to the diluted secondary antibody (ab205718) for incubation. After washing three times (10 min every time), an ECL Chemiluminescence kit (Thermo-Fisher) was used for detection. The expression levels of TLR1 and protein were normalized with GAPDH and 284 Lamin A as internal parameters.
Preparation of receptors and ligandsWith crystal structure of human TLR1 (PDB ID: 6nih) as a protein receptor, the crystal water and other small molecules in the crystal structure in the pymol1.7 program were deleted, hydrogen atoms were added and saved in the program. The two-dimensional structure of curcumin was plotted with chemdraw 15.0 and saved in cdx format. Chem3D 15.0 was loaded and the MM2 force field was adopted to minimize energy. The autodocktool1.5.6 program was loaded, charged, and assigned to the atom type. As the protein remains rigid, all the rotatable bonds of curcumin were set to be flexible and saved in pdbqt format for further analysis.
Docking analysisAutodock vina1.1.2 was applied for molecular docking. The docking parameters were set as follows: the center coordinates of the conformation search box: center_x=103.3, center_y=−35.634, center_z=16.573. Box size: size_x=70, size_y=52, size_z=54. Other parameters: exhaustiveness=100; num_modes=9; energy_range=4. The best conformation of affinity was selected as the result of molecular docking, and subsequent analysis was performed.
Statistical approachAll experimental data in this research were presented in terms of (x±sd), calculated and plotted using GraphPad 8. An independent-sample T test was applied in group comparison and one-way analysis of variance in comparison among groups, LSD-t was used for post-hoc pairwise comparison. Results with p<0.05 were considered statistically significant.
ResultsScreening and identification of differentially expressed genes in septic cardiomyocytesWe performed differential expression analysis between the CLP and Sham groups based on the GSE125042 microarray expression matrix. A total of 2876 DEGs were screened with ∣log2FC∣ >0.2630 and adj. p<0.05, of which 1424 genes were upregulated and 1452 genes were down regulated. Volcano plots of DEG expression are shown in Figure 1A, and a heat map of DEG cluster analysis is shown in Figure 1B.
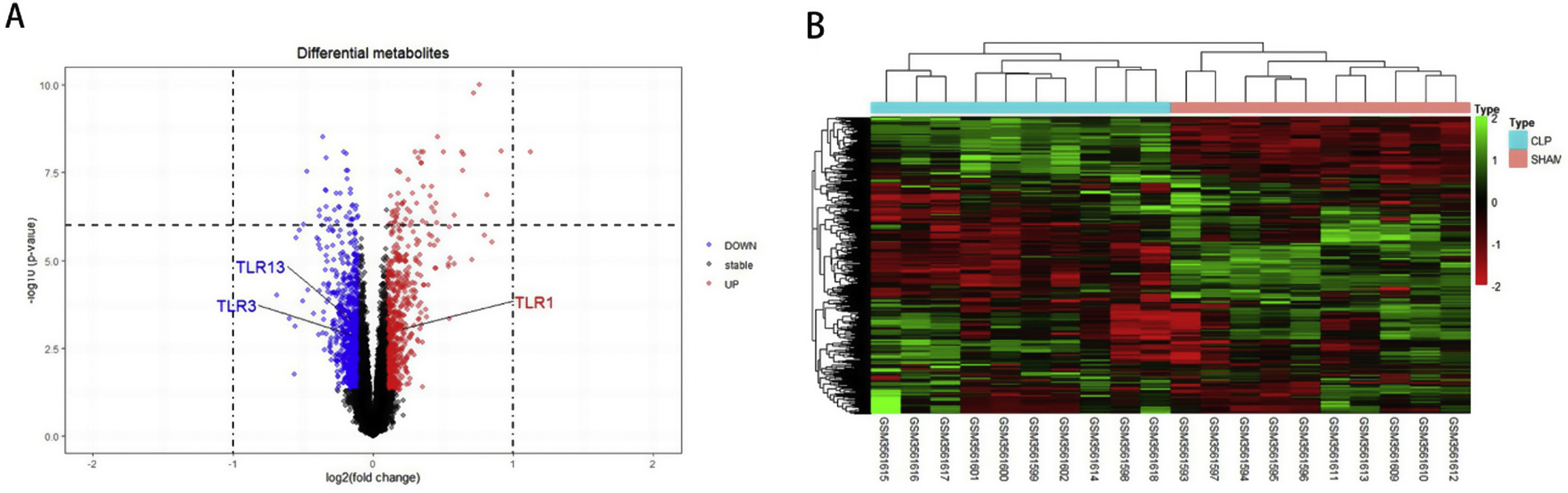
Screening and identification of differential genes for myocardial injury in sepsis. (A) Differentially expressed genes are shown as a volcano map, with red indicating up-regulated genes and blue indicating down regulated genes. (B) The expression of differential genes in each sample is represented by a heat map.
Weighted gene co-expression network analysis of whole genes based on the GSE125042 microarray expression matrix was performed to reveal genes and co-expression networks highly associated with myocardial injury in sepsis. WGCNA analysis classified genes into a total of 20 modules (Figure 2A and B), where a heat map of module-phenotype correlations showed that genes in the Brown module were most highly associated with myocardial injury in sepsis (Figure 2C and D, p<0.05, R=0.94). Interestingly, TLR1 was also present in the Brown module with the highest correlation (Table 2).
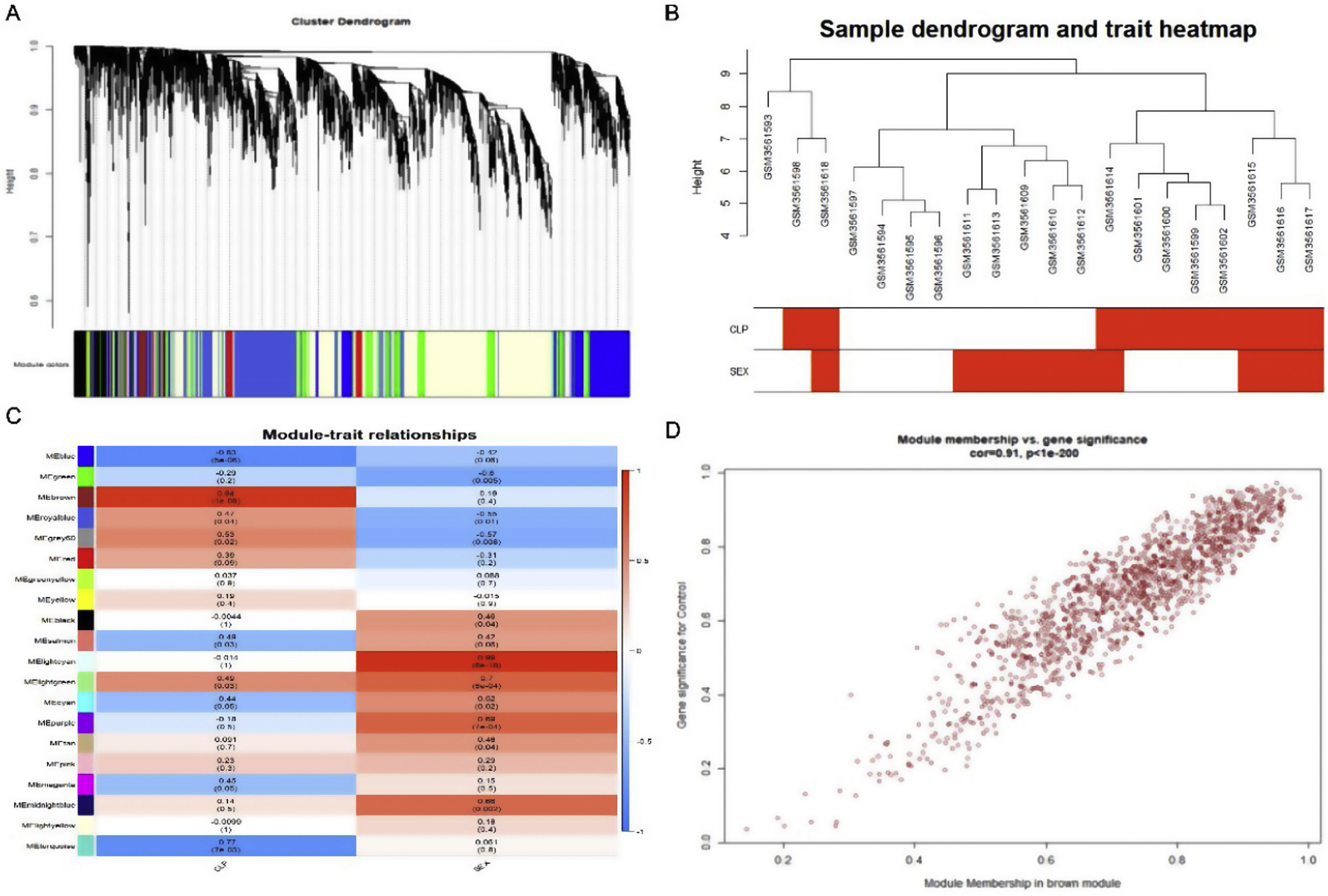
Weighted gene co-expression network analysis (WGCNA) of gene in septic cardiomyocytes. (A) Co-expression module-level cluster tree identified by WGCNA; (B) cluster tree diagram: the relationship between septic cardiomyocyte samples; (C) relationship between sepsis model and module relevance; (D) scatter diagram between members of the Brown module and genetic sense.
The Upset Venn diagram can better display the quantitative results of multiple interaction sets than a traditional Venn diagram. The common gene distribution of the up/down regulated DEGs and WGCNA modules of the highest correlation is shown in Figure 3. Since TLR1 is an up-regulated gene and located in the Brown module, the up-regulated DEGs were intersected with the Brown module of highest relevance to obtain 601 genes. According to the enrichment analysis, these 601 genes were significantly enriched in the generation of ribosomal protein complexes, ribose RNA-related metabolism, myeloblast migration and other related biological processes; in phagocytic vesicles, membrane rafts and other related cellular components; as well as in the binding of a Toll-like receptor, amide binding and other related molecular functions. According to the analysis on KEGG pathway, these 601 genes are mainly enriched in NF-κB signaling pathway, autophagy, mitochondrial autophagy and other pathways. In addition, these genes are also involved in proteasome, RNA transport, TGF-beta signaling pathway and JAK-STAT signaling pathways. The correlation can be clarified by visualizing the interactive network of sepsis-related pathways and corresponding genes. See Figure 4 and Table 3 for details.
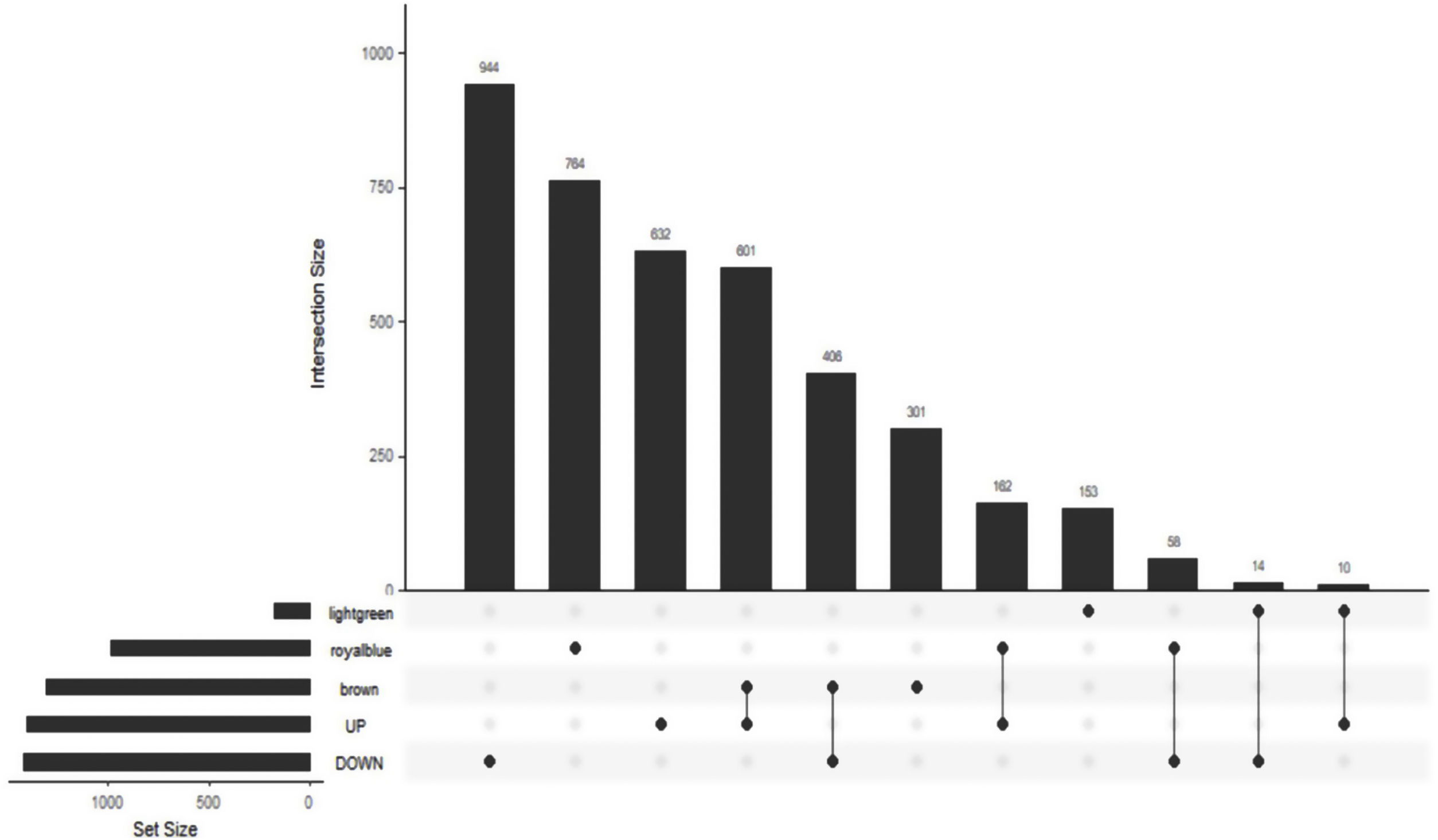
Upset Venn diagram of up/down regulated differential expression genes and gene in weighted gene co-expression network analysis modules. The histogram above represents the number of genes contained in each group, the bar in the lower left corner shows the number of genes contained in each type, and the dots and rows in the lower right corner indicate the types of events included.
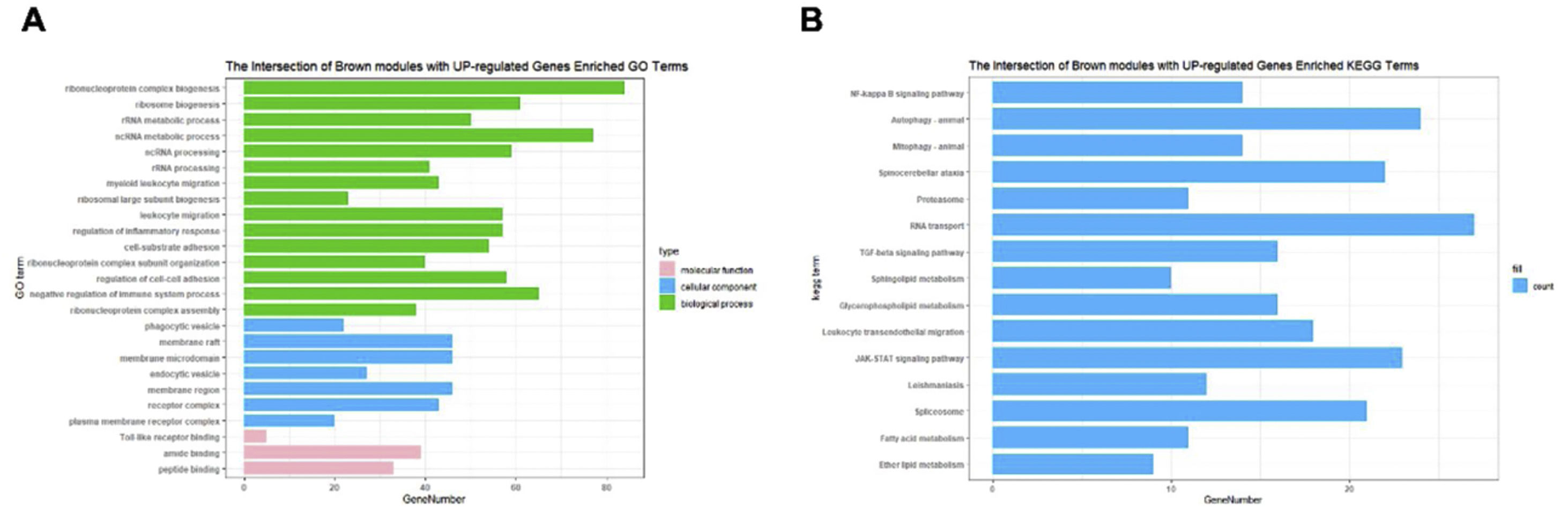
Functional enrichment analysis on the up/down regulated differential expression genes and genes in weighted gene co-expression network analysis module. (A) Annotation of the entries of 601 genes in biological process (BP), molecular function, and cell composition, of which BP takes the top 15. (B) The top 15 of Kyoto Encyclopedia of Genes and Genomes enrichment analysis.
Gene ontology enrichment analysis on the up/down-regulated DEGs and genes in WGCNA module.
| ID | Category | Description | p value | Q value | Count |
|---|---|---|---|---|---|
| GO:0022613 | BP | Ribonucleoprotein complex biogenesis | 5.33E−20 | 2.31E−16 | 84 |
| GO:0042254 | BP | Ribosome biogenesis | 3.30E−17 | 7.15E−14 | 61 |
| GO:0016072 | BP | rRNA metabolic process | 2.45E−14 | 3.54E−11 | 50 |
| GO:0034660 | BP | ncRNA metabolic process | 3.95E−14 | 4.27E−11 | 77 |
| GO:0034470 | BP | ncRNA processing | 3.27E−12 | 2.83E−09 | 59 |
| GO:0006364 | BP | rRNA processing | 1.08E−11 | 7.81E−09 | 41 |
| GO:0097529 | BP | Myeloid leukocyte migration | 1.27E−11 | 7.88E−09 | 43 |
| GO:0042273 | BP | Ribosomal large subunit biogenesis | 2.31E−11 | 1.25E−08 | 23 |
| GO:0050900 | BP | Leukocyte migration | 6.48E−11 | 3.12E−08 | 57 |
| GO:0045335 | CC | Phagocytic vesicle | 8.91E−08 | 7.50E−06 | 22 |
| GO:0045121 | CC | Membrane raft | 1.78E−05 | 0.000498792 | 46 |
| GO:0098857 | CC | Membrane microdomain | 1.90E−05 | 0.000506272 | 46 |
| GO:0030139 | CC | Endocytic vesicle | 3.63E−05 | 0.000832612 | 27 |
| GO:0098589 | CC | Membrane region | 4.23E−05 | 0.000913644 | 46 |
| GO:0043235 | CC | Receptor complex | 0.000667428 | 0.005814256 | 43 |
| GO:0098802 | CC | Plasma membrane receptor complex | 0.01481182 | 0.05846771 | 20 |
| GO:0035325 | MF | Toll-like receptor binding | 0.001440599 | 0.022689084 | 5 |
| GO:0033218 | MF | Amide binding | 0.00166233 | 0.02569644 | 39 |
| GO:0042277 | MF | Peptide binding | 0.002146686 | 0.028901902 | 33 |
We performed GSEA analysis of 601 genes obtained by taking the intersection of the upregulated DEGs with the Brown module with the highest correlation, and obtained 62 pathways. Three gene sets were statistically significant, and nine gene sets had an incidence rate of <0.25, mainly including human papillomavirus infection, chemokine signaling pathway, NF-κB signaling pathway and other related processes. The results are shown in Table 4. NF-κB signaling pathways were significantly enriched (NES=1.146, p=0.024) in sepsis-induced cardiomyocytes, with an enrichment score of 0.545. It is suggested that TLR1 may affect the survival of cardiomyocytes by interacting with the NF-κB signaling pathway, serving as a molecular intervention target for septic cardiomyopathy, see Figure 5 for details.
Gene set enrichment analysis on the up/down-regulated DEGs and genes in WGCNA module.
| ID | Description | Set size | Enrichment score | NES | p value | Rank |
|---|---|---|---|---|---|---|
| mmu05165 | Human papillomavirus infection | 24 | 0.464187592 | 1.044600419 | 0.44278607 | 141 |
| mmu04062 | Chemokine signaling pathway | 21 | 0.568588648 | 1.273292575 | 0.163265306 | 98 |
| mmu04064 | NF-κB signaling pathway | 14 | 0.545138139 | 1.146425796 | 0.024031414 | 457 |
| mmu04621 | NOD-like receptor signaling pathway | 14 | 0.600319758 | 1.26247277 | 0.20418848 | 125 |
| mmu05418 | Fluid shear stress and atherosclerosis | 18 | 0.66693923 | 1.452330629 | 0.076923077 | 28 |
| mmu04310 | Wnt signaling pathway | 18 | 0.290075989 | 0.63167111 | 0.892307692 | 278 |
| mmu04060 | Cytokine-cytokine receptor interaction | 29 | 0.67392217 | 1.511878939 | 0.039800995 | 85 |
| mmu01100 | Metabolic pathways | 133 | 0.434845227 | 1.028150515 | 0.422885572 | 294 |
| mmu04360 | Axon guidance | 17 | 0.231266649 | 0.494958121 | 0.963541667 | 410 |
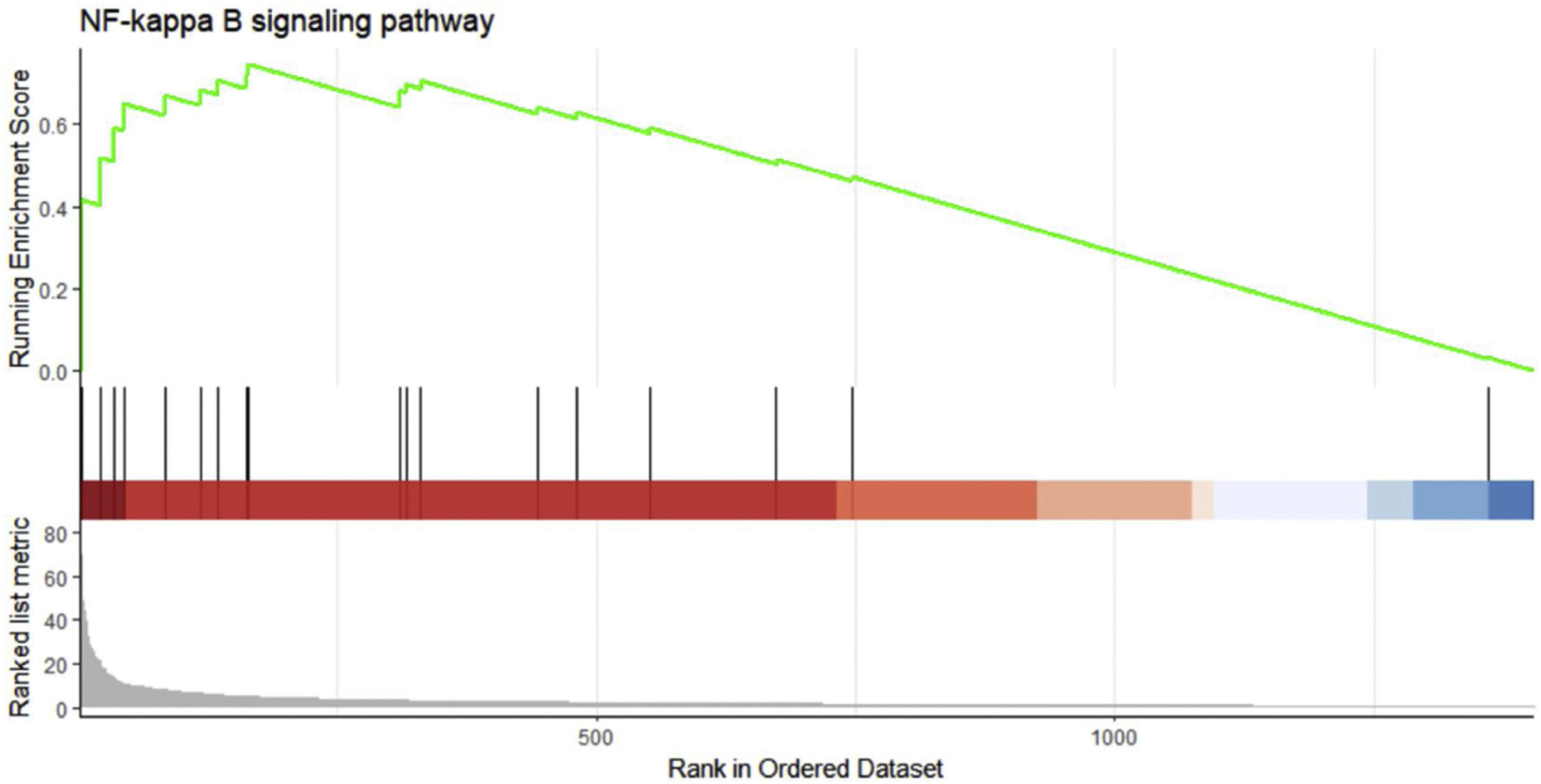
Gene set enrichment analysis on the up/down-regulated differential expression genes and genes in weighted gene co-expression network analysis module. These 601 genes were significantly enriched in the NF-κB signaling pathway, and the green curve represents the change in gene density identified in the RNA sequence.
Lipopolysaccharide was used to construct a rat model of sepsis, and the effect of intraperitoneal injection of curcumin on rats with sepsis was observed. Curcumin increased the overall survival of rats seven days after LPS induction. Blood was collected from the rats’ hearts, and the expression levels of inflammatory factors and myocardial enzymes in serum were detected. The results showed that curcumin effectively reduced the expression of pro-inflammatory factors TNF-α and IL-6, up-regulated the expression of anti-inflammatory factors IL-10, and down regulated the expression levels of BNP, cTn?, CK and CK-MB (p<0.05) (Figure 6). Then we further examined the effect of curcumin on the model of myocardial injury induced by LPS through in vitro cell experiments, we first added 0, 10, 30, 50 and 100 μM curcumin to H9C2 cells in order, then we treated the samples with LPS to induce cardiomyocyte injury after 12 hours. The QPCR showed that curcumin can inhibit the expression levels of TLR1 mRNA and NF-κB mRNA in H9C2 cells. Western blot detected that the protein expression of TLR1 and p-NF-κB was down regulated. After double staining detection with CCK-8 and Annexin/PI, it was found that curcumin can improve the survival rate of cells and inhibit apoptosis after LPS treatment, which is dose-dependent (p<0.05). See Figure 7 for details.
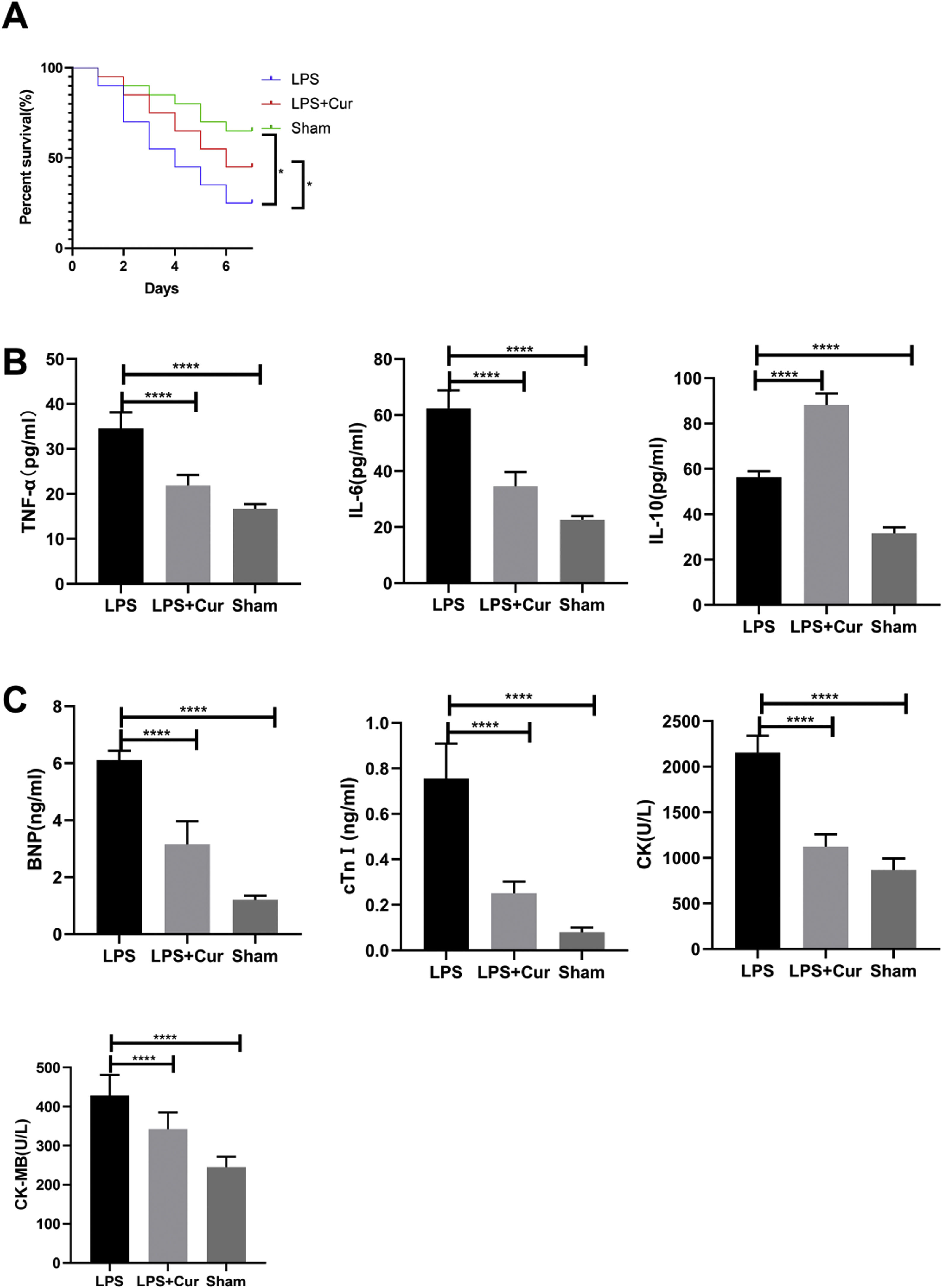
Curcumin improves myocardial damage induced by LPS in rats with sepsis. (A) 7-day survival curves of rats in each group; (B) expression levels of inflammatory factors TNF-α, IL-6 and IL-10 in serum of rats in each group; (C) expression levels of cardiac enzymes BNP, cTn, CK and CK-MB in serum of rats in each group. Note: *p<0.05, ****p<0.0001.
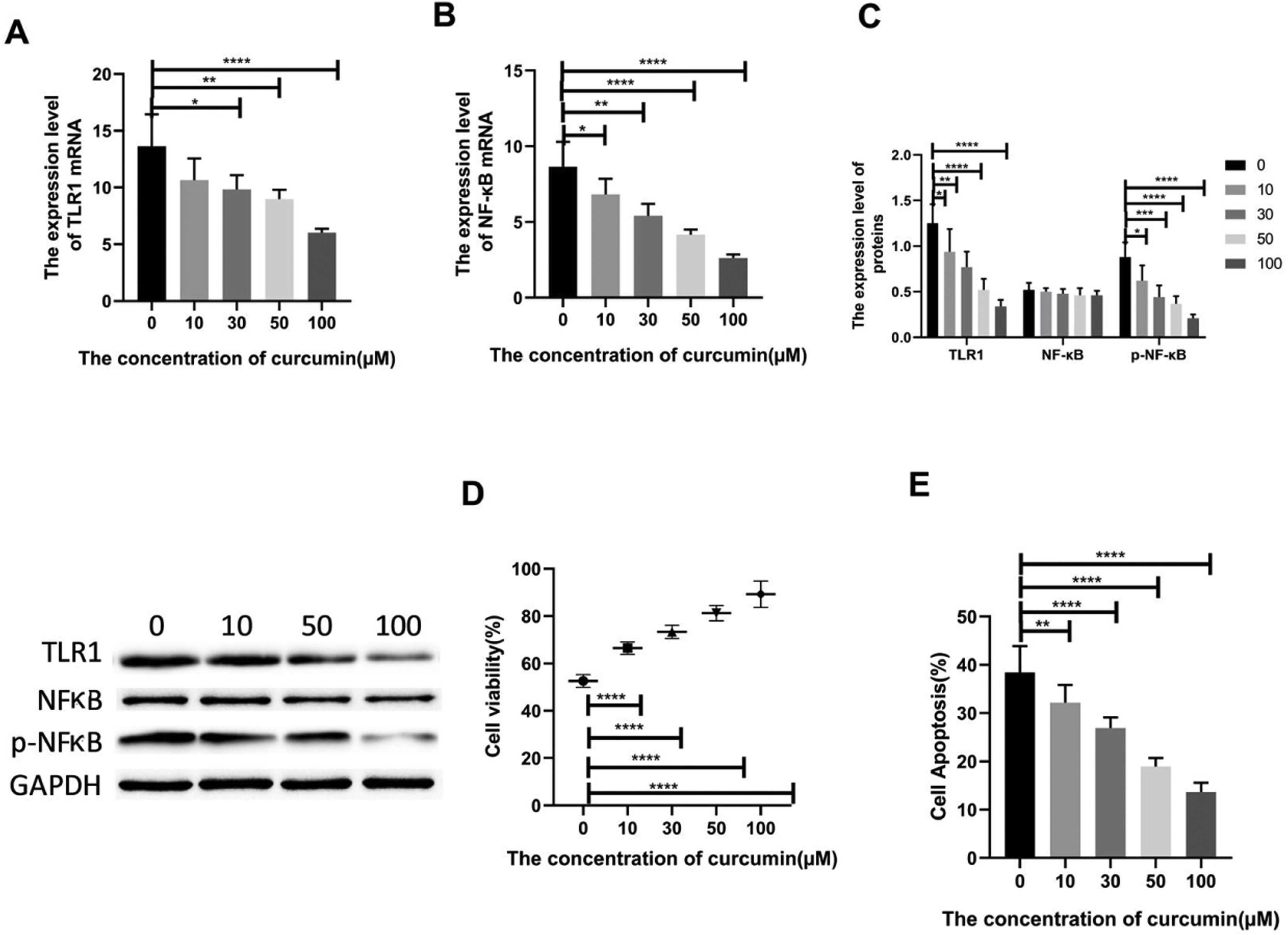
Curcumin increases the cell survival rate of LPS-treated cardiomyocytes. (A) Effect of different concentrations of curcumin on the relative expression levels of TLR1 mRNA in cells after LPS treatment of H9C2. (B) Effect of different concentrations of curcumin on the relative expression levels of NF-κB mRNA in cells after LPS treatment of H9C2. (C) WB plots showing the effect of different concentrations of curcumin on the relative expression levels of TLR1 and NF-κB protein in cells after LPS treatment of H9C2. (D) Effect of different concentrations of curcumin on the cell survival rate after LPS treatment of H9C2. (E) Effect of different concentrations of curcumin on the apoptosis rate of cells after LPS treatment of H9C2. Note: *p<0.05, **p<0.01, ****p<0.0001.
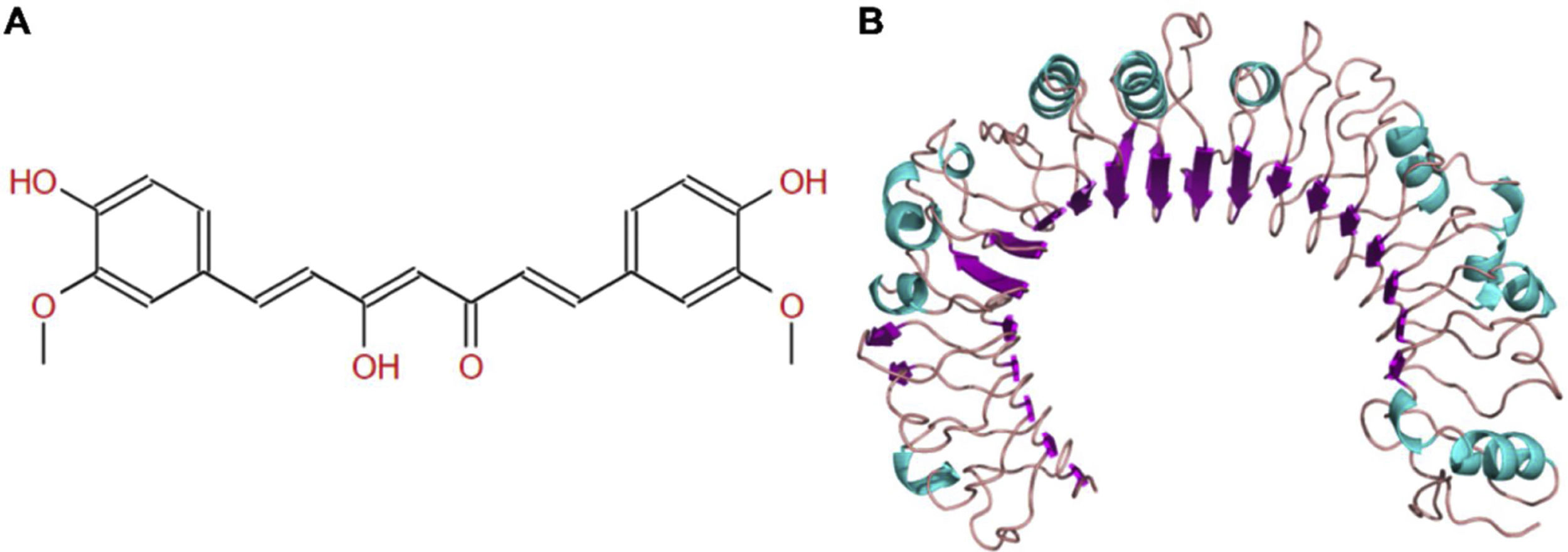
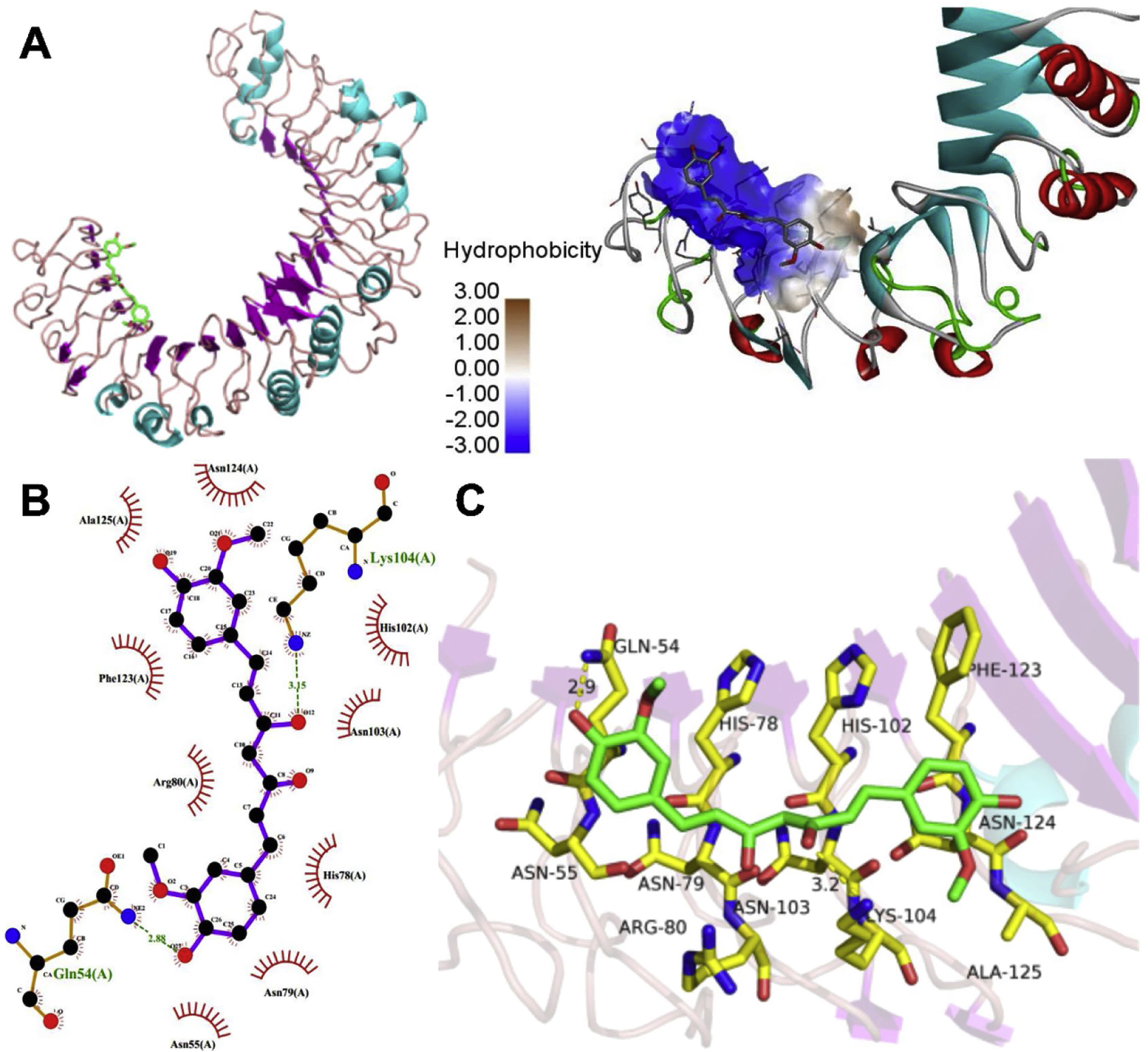
The molecular structure of curcumin and TLR1 is shown in Figure 8. Molecular docking shows that TLR1 has no obvious ligand-binding pockets or cavities. Curcumin can only bind to the area composed of multiple loops on the surface of TLR1, and the binding energy of the two can be −7.1 kcal/mol. This area has strong hydrophilicity when exposed to a solvent environment. Curcumin has multiple hydroxyl groups and oxygen, which can form hydrogen bonds with the receptor to stabilize binding (Figure 9A). It can be seen from Figure 9 that the hydroxy oxygen of curcumin (O27) can form a hydrogen bond with the side chain amino group of Gln54, and the heavy atom distance is 2.88 Å. The curcumin carbonyl oxygen (O12) can form a hydrogen bond with the side chain amino group of Lys104, with a heavy atom distance of 3.15 Å. These two hydrogen bonds are essential for the binding of curcumin. In addition, there is also a hydrophobic accumulation between Phe123 and the curcumin benzene ring, which can facilitate binding to a certain extent (Figure 9B and C).
Sepsis is known to be a complex clinical syndrome, and the specific pathogenesis of sepsis remains unclear. Therefore, study on media related to the development of sepsis is of great significance to reduce the high mortality among sepsis patients.16 DEG screening and WGCNA provide differential analysis and a functional correlation on gene expression profiles; can annotate the functions and signal pathways of these genes through enrichment analysis, which helps to better screen out the key genes in septic cardiomyopathy and further provide effective targets for clinical treatment.17–19
Johnson et al.20 conducted research on systemic inflammation and sepsis, they discovered that >80% DEGs expression was found to be upregulated. These DEGs are mostly related to the function of TLRs pathway, MAPK signaling pathway and cell apoptosis. After screening the DEGs of the chip expression matrix in the GEO database and performing WGCNA, we found that 1424 genes were upregulated in the chip expression matrix of septic cardiomyocytes. After taking an intersection from the Brown module of the highest correlation in WGCNA, we found that the intersected 601 genes were mainly enriched in MF bound by Toll-like receptors, which are involved in NF-κB signaling pathway, autophagy, mitophagy, RNA transport, TGF-beta signaling pathway, JAK-STAT and other signaling pathways. Gene set enrichment analysis (GSEA) is now an indispensable part of biological research, which can provide biological insights in high-throughput omics data.21 By performing GSEA on this gene set, we also noticed that the NF-κB signaling pathway in sepsis-induced cardiomyocytes are significantly enriched, suggesting that the NF-κB signaling pathway is highly correlated to this gene.
TLR1, an up-regulated DEG with most significant difference, also exists in the Brown module. As a result, this study sought to use TLR1 as the main molecular target in septic cardiomyocytes for further biological function analysis. TLR is a transmembrane protein with extracellular domain, containing a leucine repeated structure domain that can mediate and identify pathogen-related molecular patterns and endogenous-related molecular patterns. TLR1, TLR2, TLR4, TLR6 and TLR10 have had validated expression on cell surfaces, and TLR expression also exists in these cells as macrophages, neutrophils, natural killer cells and fibroblasts; this plays the role of activating the immune system.22,23 It has been discovered that TLR1 can form a heterodimer with TLR2 or a homodimer with itself, recognize lipoproteins of different configurations, activate MyD 88 and NF-κB or PI3K/Akt and mTOR signaling pathways, thereby regulate cell proliferation and apoptosis, participating in immune regulation in the body, therefore promoting homeostasis.24–28
Reportedly, TLRs are abnormally expressed in neonatal sepsis,29 and numerous studies have confirmed that LPS can induce myocardial damage in cells and animal models caused by sepsis.30,31 LPS can also exert a regulatory effect on the expression of TLR1, TLR2, and TLR4 in septic monocytes.29 Cardiomyocyte apoptosis can be attributed to sepsis and septic cardiomyopathy, consequently, targeted stimulation of cardiomyocytes may be an effective strategy for preventing septic cardiomyopathy.31 Interestingly, the study found that triacylated lipoprotein synergistically binding with TLR1 and TLR2 can form a stable ternary complex, which will contribute to intracellular signal transduction.32 CD14, as a receptor for LPS, can promote the endocytosis of organelles after being combined with it, thereby enhancing the pro-inflammatory properties of LPS.33 CD14 also acts as a mobile carrier for triacyl lipopeptides or lipoproteins to drive the formation of ternary complexes, greatly enhancing the sensitivity of TLRs on cell surface to lipoproteins and promoting TLR activation.32
As a natural food additive, curcumin has huge potential for anti-inflammatory and anti-oxidation treatments, furthermore, it can protect the heart by regulating oxidative stress, apoptosis and inflammation in heart diseases.34 It has been confirmed that curcumin can attenuate PI3K/AKT and CYP2E/Nrf2/ROS signal transduction, inhibit inflammation and apoptosis signals in LPS-induced mice sepsis,35 down regulate the expression of pro-inflammatory factors in cells as TNF-α and IL-6,35–37 and up regulate the expression of IL-10 anti-inflammatory factors to inhibit the inflammation of sepsis,38 which is in line with our research results. On the other hand, we found that curcumin treatment can also inhibit the expression of TLR1 and phosphorylation of NF-κB. We hypothesize that curcumin may increase the survival rate of LPS-induced cardiomyocytes by inhibiting TLR1. Choteau et al.39 confirmed that the loss of TLR1 can lead to an increase in IL-10 expression level, which supports our viewpoint that TLR1 is involved in the tissue damage caused by the inflammatory reaction in septic cardiomyocytes. In order to further clarify the inhibitory effect of curcumin on TLR1, we carried out molecular docking analysis on curcumin and TLR1. The results revealed that curcumin can form hydrogen bonds with the Gln54 and Lys104 side chain amino groups in TLR1, thus indicating that curcumin may be an inhibitor of TLR1.
In conclusion, we discovered that TLR1 displayed abnormal expression in septic myocardial injury models via chip mining, which may be a key gene for new targets for the treatment of sepsis. And for the first time, we discovered that curcumin may be used as an inhibitor of TLR1 and can effectively: improve LPS-induced myocardial injury in rats with sepsis and cardiac dysfunction; reduce inflammation, and improve the survival rate of LPS-induced cardiomyocytes, all of which deserve further research.
Ethical approvalOur research does not contain any human research or animal experiment.
FundingThis work was supported by the Key Science and Technology Project of Haikou City under grant 2015-034.
Conflicts of InterestNone declared.
