








Type IV Ehlers-Danlos syndrome (vascular) is a rare connective tissue disease caused by COL3A1 gene mutation on type III collagen. Clinical presentation is related to vascular fragility and risk of rupture of the arterial wall. Definite diagnosis is given by genetic study and the approach to these patients requires a multidisciplinary team and effective blood pressure control. There is currently only one medication with potential benefit in prevention of cardiovascular events: celiprolol. This article describes the case of a 41-year-old female patient, diagnosed with vascular Ehlers-Danlos syndrome after multiple major cardiovascular events: aortic, coronary and carotid dissections and venous and arterial thrombosis. These required multiple surgical interventions and long-term admission in intensive care units leading to complete functional recovery. This case report seeks to stress the need for an early diagnosis to prevent the severe cardiovascular complications of this rare syndrome.
A síndrome de Ehlers-Danlos tipo IV (vascular) é uma doença rara do tecido conjuntivo, caracterizada por uma mutação no gene COL3A1 do colagénio tipo III. A apresentação clínica cursa com fragilidade vascular e risco de rutura da parede arterial em idades jovens. O diagnóstico definitivo é estabelecido pelo estudo genético e a abordagem destes doentes assenta numa equipa multidisciplinar e num eficaz controlo da pressão arterial, existindo atualmente um único fármaco com aparente benefício na prevenção de eventos vasculares, o celiprolol. Este artigo descreve o caso de uma doente, cujo diagnóstico de síndrome de Ehlers-Danlos vascular foi feito aos 41 anos, na sequência múltiplos eventos cardiovasculares major, a saber: dissecção aórtica, coronária, carotídea, tromboses arteriais e venosas, que implicaram múltiplas intervenções cirúrgicas e internamento prolongado em Unidade de Cuidados Intensivos, com total recuperação funcional. Pretende-se, com este caso, salientar a gravidade das manifestações cardiovasculares desta síndrome e a necessidade do diagnóstico precoce, com o objectivo de prevenção das mesmas.
Ehlers-Danlos syndrome type IV (vascular) is a rare, inherited, autosomal dominant disease of the connective tissue, whose prevalence is estimated to be 1/100 000–1/200 000 (5% of all patients with Ehlers-Danlos syndrome), with no gender predominance.1–5 Average life expectancy is estimated to be 48 years (49 years in men and 53 years in women).4–6 However, sudden death in patients with vascular Ehlers-Danlos Syndrome can occur before the age of 20 and is more frequent in males.2
Clinical presentation ranges from patients with acrogeria (aged facial appearance), thin translucent skin with a visible subcutaneous vascular network, frequent spontaneous bruising and hematomas, to vascular ruptures in large and medium caliber arteries, potentially affecting all organs. There are also other complications that are more frequent in these patients, including perforations of the digestive tract,2 obstetric complications in the third trimester of pregnancy, spontaneous pneumothorax, and mitral valve prolapse.1,5,7
A strategy for the therapeutic approach and follow-up of patients is required to prevent complications arising from vascular fragility (including arterial dissections and aneurysmal disease). Strict control of blood pressure is, therefore, necessary, as is the involvement of a multidisciplinary team.12
Clinical caseThis article presents the case of a 41-year-old Caucasian female who is a self-employed financial analyst.
The following aspects of her medical history are relevant to her current illness:
- -
inguinal hernia surgery at the age of 5;
- -
endovascular treatment of a carotid-cavernous fistula in 2002, which initially presented as diplopia and decreased visual acuity on the right side; this procedure was complicated by dissection of the external iliac artery and the right common femoral artery;
- -
bilateral varicose veins surgery at the age of 22;
- -
unspecified intra-abdominal hemorrhage in 2016 for which she underwent conservative treatment.
Her medical history also includes spontaneous ecchymoses in childhood, ability to perform joint movements of large amplitude and difficulty in closing her eyelids while sleeping. There is no reference to a diagnosis of hypertension and her obstetric history mentions only one pregnancy without complications. Usual medication included acetylsalicylic acid 100 mg, bioflavonoids and an oral contraceptive.
The history of the current disease began in March 2018, during a work meeting in France, when the patient reported precordial pain with when the patient reported precordial. The electrocardiogram initially performed showed no ST-segment elevation and the maximum troponin value was 235 ng/L. A coronary angiography was performed at Chambérry Hospital, which also showed no significant lesions. The patient was treated with acetylsalicylic acid 100 mg, atorvastatin 10 mg, bisoprolol 2.5 mg and ramipril 1.25 mg, Figure 1

Three days later, she experienced a new presentation of typical precordial pain, resistant to sublingual nitrate therapy. The electrocardiogram documented acute myocardial infarction of the anterolateral wall with supra-ST-segment elevation (Figure 2).

A repeat coronary angiography was performed, which showed a thrombosis in myocardial infarction (TIMI 2) subocclusive lesion in the anterior descending artery (90–99%). Here an anti-proliferative drug-coated stent (BioMatrix Flex 3.5×14 mm) was placed with a TIMI 3 final result (Figure 3). During the procedure, aortography was performed, which did not reveal any suspect image of aortic dissection.

The echocardiogram subsequently performed showed a left aorto-atrial fistula located 3 mm from the aortic annulus in the non-coronary sinus next to the left commissure, aortic insufficiency grade 2/4, with valve gradient between 5-6 mmHg and pericardial effusion of 6 mm without hemodynamic compromise. Additionally, with reference to preserved global systolic function (50%), the exam revealed no dilatation of the right cavities (RA-RV gradient 20 mmHg), with mitral Doppler flow type III. Due to evidence of double contour imaging of the aortic wall, a computed tomography angiogram was performed which identified a type A aortic dissection and dissection of the celiac trunk extending to the common hepatic artery (Figure 4). The patient was then transferred to Grenoble Hospital Center, where she underwent surgery with placement of an aorto-aortic prosthesis, aortic valve replacement with a Carpentier bioprosthesis, and fistula closure. The echocardiogram performed after surgery confirmed good global systolic function, a small periprosthetic leak, and minimal mitral insufficiency, with no dilatation of the right cavities.

Over the subsequent months (March to June 2017), the patient remained in the intensive care unit, following several complications (Tables 1 and 2). Evolution was initially unfavorable, characterized by multiple organ dysfunction, requiring life support, including, cardiovascular (vasopressor support with noradrenaline), respiratory (invasive mechanical ventilation) and hematologic (disseminated intravascular coagulation criteria not met, but requiring massive transfusion) support.
Main vascular complications.
| Major Vascular Complications |
|---|
| • Aortic dissection type A (with subocclusive lesion of the anterior descending coronary artery) |
| • Dissection of the celiac trunk (with extension to the common hepatic artery) |
| • Occlusive dissection of the right internal carotid artery (with ischemia in right middle cerebral artery region) |
| • Jugular, iliac and inferior vena cava vein thrombosis |
| • Intestinal ischemia. |
Main infectious complications and isolated microorganisms.
| Main Infectious Complications And Microorganisms | |
|---|---|
| Bacteremia | Streptococcus |
| Ventilator-associated pneumonia | E. coli, H. influenzae, Enterococcusfaecalis |
| Empyema of left lower lobe | Staphilococcusepidermidis |
| Bilateral keratitis | |
| Corneal ulcer | |
Neurologically, after reducing sedation and analgesia, left hemiplegia was observed as a result of vascular ischemia in the right middle cerebral artery region, secondary to occlusive dissection of the right internal carotid artery, confirmed by Doppler.
There were also multiple pro-thrombotic arterial and venous complications (which were subsequently resolved), the most notable of which were jugular, iliac, and inferior vena cava thrombosis.
Also, during hospitalization, after an episode of upper digestive hemorrhage, the existence of right colon ischemia was confirmed.
Difficulty in weaning the patient from the ventilator remained throughout hospitalization, due to multiple complications, including bilateral pneumothorax (Figure 5) requiring drainage; on the right, complicated by severe acute respiratory distress syndrome and evolution to hemorrhagic shock, with hemothorax requiring hemostasis surgery. Due to the continued need for invasive ventilation, a tracheostomy was performed in May 2017.

We also highlight several infectious complications, requiring multiple courses of targeted antibiotic therapy (Table 2).
Once her vital functions had been stabilized, the patient was transferred to the intensive care unit of Santa Maria Hospital in 06/2017, even though she still required invasive mechanical ventilation
When considering a possible diagnosis, a collagen disease associated with vascular fragility was hypothesized, ruling out infectious and autoimmune causes, especially anti-phospholipid antibody syndrome. The study of hereditary and acquired thrombophilia was also negative. Phenotypically, there was prominence of the eyeballs, a narrow nose and thin upper lip.
After mechanical ventilation was discontinued, the patient was transferred to the Medicine I-C Department, where she underwent further diagnostic study, including a genetic study, which subsequently identified a pathogenic mutation in heterozygosity in the de novo (not identified in either parent), COL3A1 gene confirming Ehlers-Danlos syndrome type IV (Figure 6).

In collaboration with the Paris Center for Rare Vascular Diseases, the following protocol was established for the approach and follow-up of this patient:
- •
suspension of anticoagulation with enoxaparin after confirming the absence of intracardiac thrombi;
- •
maintenance of antiplatelets with acetylsalicylic acid 100 mg id, with laboratory blood testing every six months;
- •
Annual evaluation (or earlier if warranted) with Doppler echocardiography of the neck and transcranial vessels, chest, abdominal and pelvic CTA, and transthoracic echocardiography;
- •
strict control of blood pressure with celiprolol (the only beta blocker (BB) with demonstrated benefit in this disease)12,13 at the maximum tolerated dose (currently 100 mg bid), for target systolic blood pressure values of maximum 120 mmHg.
Approximately one month after admission to Santa Maria Hospital, the patient was discharged home, totally autonomous for daily living activities, maintaining respiratory kinesiotherapy and motor rehabilitation, with almost complete resolution of the left hemiparesis (brachial predominance at the time). The patient was referred to Internal Medicine, Otorhinolaryngology and Corneal Disorders, where she continued to be followed.
DiscussionThe natural history of this disease begins with hip dislocations and equinovarus feet. Also ecchymoses, thin translucent skin and a characteristic face can be identified in childhood.2 Later, in adolescence or adulthood, intestinal perforation and recurrent spontaneous pneumothorax may point to this diagnosis. Unlike the other subtypes of Ehlers-Danlos Syndrome, in type IV skin hyperextensibility and hypermobile joints are rare.7
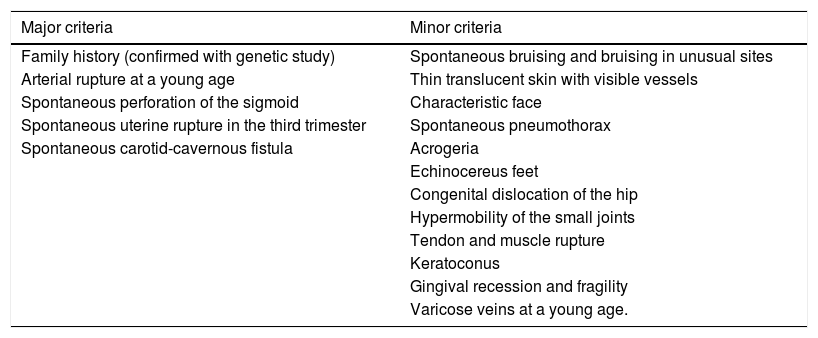
The new international classification, from 2017, led to the recognition of 13 subtypes of Ehlers-Danlos Syndromebased on clinical features.8 For each of the subtypes, there are major and minor criteria. It is possible to group them, however, the definitive diagnosis always requires confirmation via genetic testing, with the exception of the hypermobile form.8 In the case of the vascular subtype (type IV), the identification of heterozygous mutations in the COL3A1 gene (located on the long arm of chromosome 2, at position 2q24.3-q31), responsible for the deficit in collagen type III that comprises part of the arterial wall, confirms the diagnosis.1,2,5,7,10 The following table (Table 3) shows the major and minor criteria, according to the classification currently in force.8
Major and minor criteria for vascular Ehlers-Danlos syndrome.8
| Major criteria | Minor criteria |
|---|---|
| Family history (confirmed with genetic study) | Spontaneous bruising and bruising in unusual sites |
| Arterial rupture at a young age | Thin translucent skin with visible vessels |
| Spontaneous perforation of the sigmoid | Characteristic face |
| Spontaneous uterine rupture in the third trimester | Spontaneous pneumothorax |
| Spontaneous carotid-cavernous fistula | Acrogeria |
| Echinocereus feet | |
| Congenital dislocation of the hip | |
| Hypermobility of the small joints | |
| Tendon and muscle rupture | |
| Keratoconus | |
| Gingival recession and fragility | |
| Varicose veins at a young age. |
The differential diagnosis of this syndrome should include other subtypes of Ehlers-Danlos syndrome and other connective tissue diseases, such as Loeys-Dietz and Marfan syndromes. In addition to genetic testing, some phenotypic features may enable differentiation between subtyoes.2,4,9,10 Since these patients also present with arterial and venous thrombotic complications, antiphospholipid antibody syndrome should also be excluded.11,14
The clinical case presented herein demonstrates that despite the serious associated complications, this disease is often only diagnosed in adulthood. This patient, despite several early indicators of collagen disease, was not diagnosed with it until she presented major vascular events (Table 1).
Since this is a disease of autosomal dominant transmission, early diagnosis and the conduct of genetic studies in direct relatives is important. Although there is no cure for this syndrome, the prevention of major complications of the disease is possible, with the control of cardiovascular risk factors, such as hypertension.
As a rare disease, studies on treatment are scarce, and there is little consensus on how to monitor the patient and how often to perform imaging studies.
Currently, there is evidence of the use of only one BB with a particular action on TGF-beta: celiprolol.4,12 This drug, already approved by the Food and Drug Administration, has shown an apparent benefit on the mortality of patients with vascular Ehlers-Danlos syndrome.13,14 Although it is not available in Portugal, it was authorized by INFARMED in this patient's case, because there was a formal indication for its use in this disease. However, it is unknown whether other types of BBs could have the same effect on collagen in the vascular wall, as they have not been studied.
Other studies on the potential use of angiotensin receptor antagonists (ARCADE study) are currently underway.15 There also seems to be evidence that C-reactive protein may be a marker of disease activity.16
However, the following has been established:
- •
The definitive diagnosis of this syndrome has to be made by means of a genetic study, with subsequent extension to all direct relatives;
- •
Blood pressure control, ideally with systolic blood pressure values <120 mmHg, is the most important preventive measure, using celiprolol (at a maximum dose of 200 mg bid), the only drug approved so far with proven benefit in this disease;
- •
There is a need for a multidisciplinary team that periodically monitors the clinical status of these patients, with annual arterial Doppler echocardiography of the supra-aortic trunks, abdominal aorta and lower limb arteries, as well as transthoracic echocardiography. Annual thoracic-abdominal-pelvic CTA is also recommended and, in specific cases, ear, nose and throat and gynecological evaluation.
In the future, more studies will be needed to document the efficacy of other drugs or markers, always from a perspective of preventing major vascular events. The high level of suspicion needed to diagnose this disease should also be emphasized, especially when the initial presentation is frustrating and nonspecific.
In this clinical case, the patient's evolution has been favorable. She has recovered from all organ dysfunctions and has resumed her usual working activity. Mild dysphonia still persists, as a result of prolonged mechanical ventilation, and she is currently undergoing speech therapy. She also intends to undergo right vocal cord medialization. A genetic study of the parents and the daughter was performed, which proved to be negative for the mutation, thus showing that this patient is a case of de novo mutation. She continues to be followed in Internal Medicine, with rigorous blood pressure control with celiprolol 100 mg 2×/day, quarterly clinical evaluations, and annual imaging control.
Conflicts of interestThe authors have no conflicts of interest to declare.
