





Anomalous origin of the left coronary artery from the pulmonary artery is a rare congenital heart defect and a cause of myocardial ischemia during childhood. Most undiagnosed cases die in the first year of life as an extensive collateral network is essential for survival. The diagnosis requires a high index of clinical suspicion. The authors present the case of an asymptomatic eight-year-old black girl referred from Cape Verde in order to clarify left ventricular dilatation and dysfunction with turbulent systolic and diastolic flows observed at the interventricular septum. At the age of three months, she was diagnosed with heart failure, subsequently classified as dilated cardiomyopathy. She improved clinically following anticongestive therapy, which was ongoing at the time of admission to our center. The echocardiogram suggested anomalous origin of the left coronary artery from the pulmonary artery, and the diagnosis was confirmed by computed tomography angiography and cardiac catheterization. The patient underwent successful direct implantation of the left coronary artery into the aorta, creating a dual coronary perfusion system. This case illustrates an unusual presentation of a rare heart condition that survived without a diagnosis after the first year of life. It also highlights the importance of multimodality imaging in these cases.
A Origem Anómala da Artéria Coronária Esquerda da Artéria Pulmonar é uma cardiopatia congénita rara e uma causa de isquemia miocárdica em idade pediátrica. A maioria dos casos não diagnosticados morre no primeiro ano de vida, sendo necessária a formação de uma extensa rede de colaterais para permitir a sobrevivência. O diagnóstico não é linear exigindo elevada suspeição clínica. Os autores apresentam o caso de uma criança de oito anos de idade, de raça negra, assintomática, referenciada de Cabo Verde para esclarecimento de dilatação e disfunção do ventrículo esquerdo com fluxos turbulentos sistodiastólicos a nível do septo interventricular. Dos antecedentes destacava-se quadro de insuficiência cardíaca diagnosticada aos três meses de idade, com avaliação posterior compatível com miocardiopatia dilatada. Teve melhoria clínica após início de terapêutica anticongestiva que mantinha na altura da admissão no nosso Centro. Ecocardiograficamente suspeitou-se de Origem Anómala da Artéria Coronária Esquerda da Artéria Pulmonar, sendo este diagnóstico confirmado através de angiotomografia computorizada e cateterismo cardíaco. A doente foi submetida, com sucesso, a implantação direta da artéria coronária esquerda na aorta permitindo criar um sistema de perfusão coronário duplo. Este caso ilustra uma forma incomum de uma patologia rara que sobreviveu sem diagnóstico após o primeiro ano de vida. Reforça igualmente a importância da multimodalidade de imagem nestes casos.
Anomalous origin of the left coronary artery (LCA) from the pulmonary artery (ALCAPA) accounts for 0.24-0.46% of congenital heart defects.1 Although rare, it is the most common congenital coronary artery anomaly. Without treatment, only 10-15% of cases reach adulthood; survival depends on the development of an intercoronary collateral network.2
Echocardiography is the first-line diagnostic method, but other imaging exams may be required to obtain better definition of the coronary anatomy. Computed tomography angiography (CTA) can identify the origin of the coronary arteries and their relation with adjacent structures, and these measurements are crucial to planning the surgical approach. However, for many authors, selective coronary angiography remains the gold standard for surgical planning.
Surgery is the treatment of choice for patients with ALCAPA and in severely symptomatic children may have to be performed on an emergency basis. Ideally a dual coronary perfusion system should be created. In infants when the anatomy is favorable, the best option is for the most anatomical correction, the direct implantation of the LCA into the aorta. Alternatively, an intrapulmonary artery baffle can be created from the left coronary ostium to the aorta (Takeuchi procedure). However, this solution is subject to late complications including pulmonary stenosis, intrapulmonary leak, and aortic stenosis and regurgitation.3
In adults, coronary artery bypass grafting of the LCA is usually performed following ligation of its origin in the pulmonary artery. The advantage of this technique is that it avoids manipulation of the aortic root, but this must be weighed against the risk associated with a less durable graft.
The clinical course following surgical correction is generally favorable.
Case reportThe authors present the case of an eight-year-old black girl transferred from Cape Verde in order to clarify heart disease.
She had been suffering from heart failure since the age of three months, and at 11 months was diagnosed by echocardiography with dilated cardiomyopathy. Anticongestive therapy was begun, resulting in clinical improvement.
She was transferred at the age of eight years to our center for further diagnostic clarification and treatment. Although asymptomatic, she presented a continuous murmur at the left upper parasternal border.
The chest X-ray revealed mild cardiomegaly and the electrocardiogram showed criteria for left ventricular hypertrophy, with signs of subendocardial ischemia of the lateral and septal wall but no evidence of necrosis.
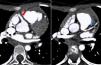
Echocardiography revealed dilatation of the left ventricle with a hypokinetic and aneurysmal apical region, and increased echogenicity of the subvalvular mitral apparatus compatible with fibroelastosis secondary to ischemia of the papillary muscle. Doppler study identified multiple interventricular systolic and diastolic flows with maximum velocity <1 ms (Figure 1A and B). There was no mitral regurgitation and ejection fraction by Simpson's method was 48%. Right coronary artery (RCA) ectasia was observed (Figure 1C) and the origin of the LCA could not be identified. Retrograde flow in the main pulmonary artery (MPA) immediately above the pulmonary valve was seen in short-axis parasternal view, suggestive of runoff from the LCA (Figure 1D, arrow).
Transthoracic echocardiogram with color Doppler. Short-axis and long-axis parasternal views show interventricular systolic and diastolic flows due to the extensive collateral network (A and B); short-axis parasternal view reveals anterograde flow in the right coronary artery, which is dilated in its proximal portion (C), and retrograde flow in the main pulmonary artery (arrow), corresponding to runoff from the left coronary artery (D).
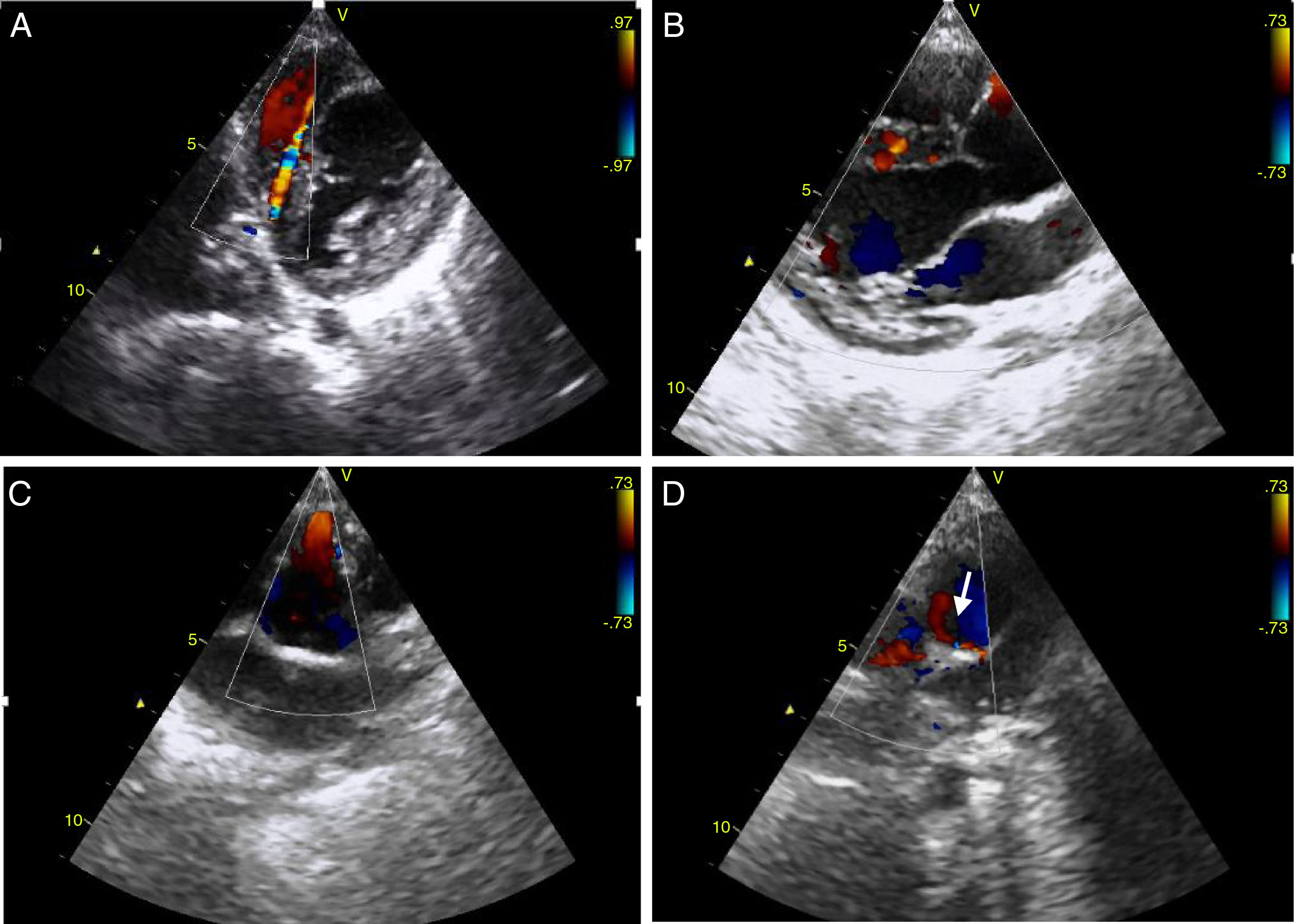
To confirm the diagnosis, CTA was performed, which showed the left main coronary artery originating from the left lower aspect of the MPA, about 10 mm from the aortic root (Figures 2 and 3). This distance raised doubts concerning the feasibility of direct implantation of the LCA into the aorta. The patient's periods of higher heart rate despite negative chronotropic medication and motion-induced artefacts meant that only the proximal portions of the coronary arteries could be defined.

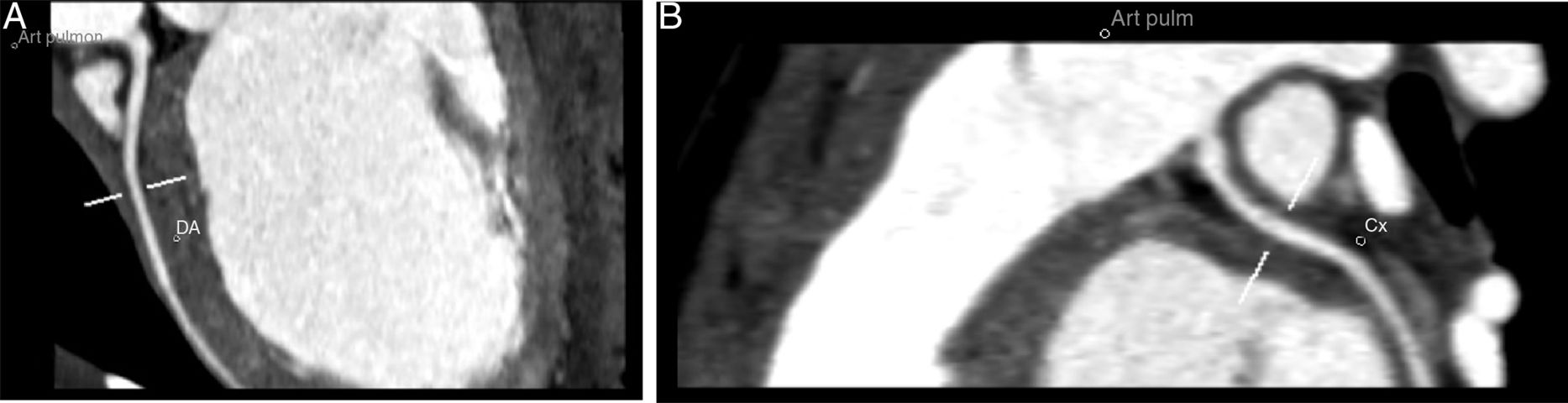
64-slice ECG-gated computed tomography, curved multiplanar reformatting, showing the left main coronary artery originating from the lower left aspect of the main pulmonary artery. The left main coronary artery has a caliber of 2.6 mm and a length of around 3 mm, with regular contours and preserved patency. It presents a normal bifurcation into the anterior descending artery (A) and circumflex artery (non-dominant) (B). These vessels have preserved patency in their visible portions (proximal half).
Cardiac catheterization confirmed the diagnosis of ALCAPA as well as retrograde perfusion of the LCE by the RCA via an extensive collateral network, followed by drainage into the MPA. It also detected reduced luminal diameter of the distal portion of the anterior descending artery (Figure 4A and C). Left ventriculography showed aneurysmal dilatation of the left ventricular apical region (Figure 4A) and mild systolic dysfunction. Left ventricular end-diastolic pressure was elevated but pulmonary vascular pressure and resistance were normal. Attempted selective catheterization of the LCA via the MPA was unsuccessful, and angiography of the MPA showed no opacification of the LCA.
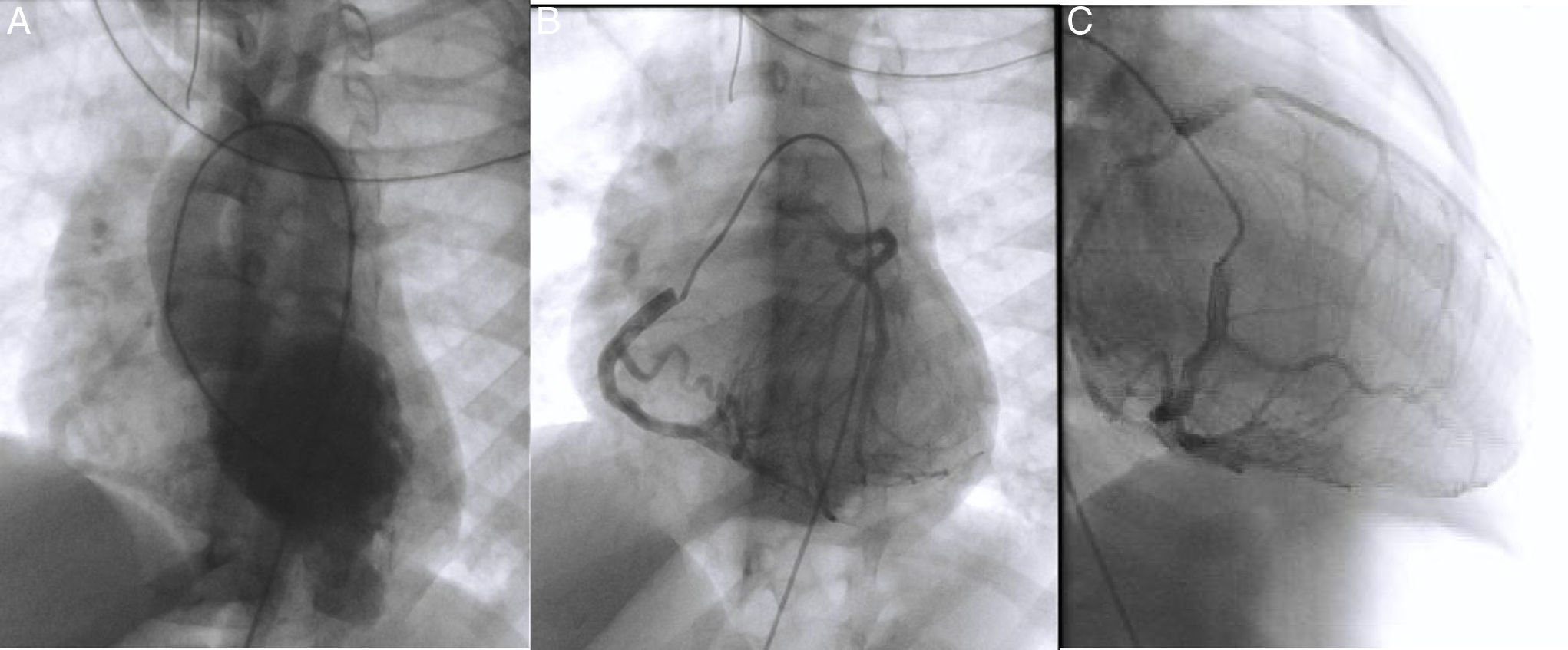
Diagnostic cardiac catheterization. Left ventriculography in posteroanterior view shows an aneurysmal region of the left ventricular apex and filling of the aorta and the right coronary artery (A); right coronary angiography in posteroanterior and right anterior oblique views shows a dilated coronary artery communicating with and filling the left coronary artery via an extensive collateral network (B and C). The left main coronary artery originates from the main pulmonary artery, and the blood flow is from aorta to right coronary artery, to collaterals, to left coronary artery, to pulmonary artery. The distal portion of the anterior descending artery is poorly filled.
Following discussion with the surgical team, the patient underwent successful direct implantation of the LCA into the aorta. Her postoperative course was favorable.
The electrocardiographic alterations observed at admission persisted. Before return to her country of origin, in order to exclude perfusion deficits following coronary implantation, she underwent exercise testing according to the Bruce protocol, which excluded arrhythmia or worsening of septal and lateral wall repolarization abnormalities with exercise. Echocardiographic assessment showed adequate flow in the reimplanted coronary artery and normal left chamber dimensions, with improvement in left ventricular systolic dysfunction.
DiscussionALCAPA can be classified into two types, infant and adult, according to its clinical expression. In fetal life, elevated pulmonary pressures lead to antegrade flow in the LCA, enabling good adaptation in utero. Following birth, the gradual lowering of pulmonary pressure and vascular resistance results in progressively reduced coronary perfusion, and eventually to flow reversal in the LCA.
In the more common infant type, a collateral network does not form after flow reversal in the LCA and the diameter of the coronary arteries remains normal. The reduced blood supply to the myocardium in the LCA territory results in ischemic heart disease that leads to 90% mortality in the first year of life.
Flow reversal in the LCA in the adult type triggers the formation of an intercoronary collateral circulation and gradual dilatation of the coronaries, enabling adequate filling pressure in the LCA from the RCA. Progressive coronary dilatation means that blood flows preferentially to the pulmonary artery rather than to the high-resistance myocardial system, leading to the phenomenon known as coronary steal. These patients develop chronic ischemic heart disease and are at high risk for sudden death due to arrhythmias.
It should be borne in mind that ventricular function depends more on perfusion pressure in the coronaries than on the oxygen saturation of the blood they transport. Thus, unlike in ALCAPA, in simple transposition of the great arteries, which is characterized by low oxygen saturation in the coronaries but normal coronary perfusion pressure, left ventricular function is preserved.
In the case presented, a child with ALCAPA survived due to the development of an extensive right-to-left coronary collateral circulation. The onset of symptoms at the age of three months may have coincided with the progressive fall in pulmonary vascular resistance and closure of the ductus arteriosus. In her country of origin, she had been diagnosed echocardiographically with dilated cardiomyopathy of uncertain cause. The diagnosis of ALCAPA does in fact require a high index of clinical suspicion and a range of diagnostic exams.
The improvement in ventricular function was probably due to the extensive collateral network that developed during the most critical phase, maintaining adequate coronary perfusion pressure and oxygen supply to the at-risk myocardium.
On admission to our center, the patient was asymptomatic, reflecting the good adaptation provided by natural revascularization. The continuous murmur heard on auscultation may have been caused by runoff from the LCA to the MPA. Echocardiography revealed the typical alterations of the adult type of ALCAPA: intraventricular systolic and diastolic flows (due to the collateral network supplying the LCA); left ventricular dilatation and dysfunction secondary to myocardial ischemia; and the LCA not originating from the aorta. However, false images of the LCA emerging from the aorta have been reported. In such cases the anomalous retrograde direction of flow in the LCA indicates the correct diagnosis.
CTA is hampered by technical difficulties at pediatric ages, and so investigation may need to be supplemented with coronary angiography, as in our patient.
Cardiac catheterization was used in this case to characterize coronary flow and the entire coronary anatomy. It demonstrated reduced luminal diameter in the distal portion of the anterior descending artery leading to inadequate perfusion in its distal territory, with an aneurysmal region and apical hypokinesis.
Surgical correction should aim to establish a dual coronary perfusion system, and this was the option chosen by our center's surgical team.
Following intervention, left ventricular dysfunction and dilatation can be expected to improve. In the case reported, although postoperative follow-up has been short, normalization of left ventricular dimensions and improvement in systolic function have been observed.
ConclusionALCAPA is a rare heart defect associated with high mortality. The natural history of the few cases that have survived un-diagnosed after the first year of life includes chronic ischemia, ventricular dysfunction, and potentially lethal arrhythmias. Multimodality diagnostic exams are important to obtain an accurate diagnosis and to provide essential information to guide surgery. Surgical correction should be considered in all patients, with preference for the establishment of a dual coronary perfusion system. Prognosis after surgery is generally favorable.
Conflicts of interestThe authors have no conflicts of interest to declare.
The authors thank Dr. Rui Catarino for his assistance in the production and choice of the images used in this case study.
Please cite this article as: Amaral ME, Epifânio P, Noronha N, Pires A, Martins P, Azevedo V, et al. Causa rara de disfunção ventricular esquerda em idade pediátrica. Rev Port Cardiol. 2019;38:159.
