






Endothelin‐1 antagonists are increasingly used in the treatment of pulmonary hypertension despite the lack of knowledge of their myocardial and systemic effects. We assessed the right ventricular myocardial and systemic effects of endothelin‐1 antagonists in monocrotaline‐induced pulmonary hypertension.
MethodsMale Wistar rats (180–200 g, n=57) randomly received 60 mg/kg monocrotaline or vehicle subcutaneously. Two days later, bosentan was randomly started (300 mg/kg/day) by oral route in a subgroup of monocrotaline‐injected rats, while the other monocrotaline‐injected and control rats received vehicle. At 25–30 days, invasive hemodynamic assessment was performed under anesthesia, arterial blood samples were collected for gas analysis and plasma was extracted for quantification of endothelin‐1, cytokines, nitrates and 6‐keto‐prostaglandin F1α. Right ventricular myocardium was collected for assessment of cyclooxygenase and nitric oxide synthase activity and gene expression.
ResultsThe monocrotaline group developed pulmonary hypertension, low cardiac output, right ventricular hypertrophy and dilation, changes in gene expression and inflammatory activation that were attenuated in the group treated with bosentan. From a functional point of view, this group had improved right ventricular function and preserved ventriculo‐vascular coupling, without deterioration in arterial gas parameters or systemic hypotension. In molecular terms, they showed reduced endothelin‐1 and cytokine levels, decreased right ventricular inducible nitric oxide synthase and cyclooxygenase‐2 activity and increased nitrate plasma levels compared with the non‐treated group.
ConclusionsIn this study we demonstrate that besides attenuating pulmonary hypertension, bosentan has beneficial hemodynamic, myocardial and anti‐inflammatory effects.
Os efeitos miocárdicos e sistémicos dos antagonistas da endotelina‐1 na hipertensão pulmonar são ainda pouco conhecidos. Procurámos avaliar os efeitos miocárdicos ventriculares direitos e sistémicos, no que concerne à ativação inflamatória, dos antagonistas da endotelina‐1 no modelo de hipertensão pulmonar induzida pela monocrotalina.
MétodosRatos Wistar machos (180‐200 g, n=57) receberam aleatoriamente 60 mg/kg de monocrotalina ou veículo, via subcutânea. Um subgrupo aleatório destes animais passou a receber bosentan 300 mg/kg/dia por via oral, dois dias após, enquanto os restantes animais do grupo monocrotalina e o grupo controlo receberam veículo. Aos 25‐30 dias procedeu‐se à avaliação hemodinâmica invasiva, colheita de sangue arterial, de plasma para quantificação de endotelina‐1, citoquinas, nitratos e 6‐ceto‐prostaglandina F1α, bem como de ventrículo direito para avaliação génica e da atividade das ciclo‐oxigénases e síntases do óxido nítrico.
ResultadosO grupo monocrotalina desenvolveu hipertensão pulmonar, dilatação e hipertrofia ventricular direita, bem como diminuição do débito cardíaco, alterações da expressão génica ventricular direita e ativação inflamatória, que foram atenuadas no grupo monocrotalina tratados com bosentan. Do ponto de vista funcional, salienta‐se que este grupo apresentou melhoria da função ventricular direita com preservação do acoplamento ventrículo‐vascular, sem deterioração da gasometria ou hipotensão sistémica e, em termos moleculares, diminuição dos níveis plasmáticos e da expressão ventricular direita de endotelina‐1 e citocinas, diminuição da atividade das síntases do óxido nítrico induzível e da ciclo‐oxigénase‐2.
ConclusãoDemonstrámos que o bosentan, para além de atenuar a hipertensão pulmonar, pode ter efeitos hemodinâmicos benéficos por ação anti‐inflamatória, protetora, no miocárdio ventricular direito.
6‐keto‐prostaglandin F1α
angiotensin‐converting enzyme
arbitrary units
B‐type natriuretic peptide
cardiac output
cyclooxygenase
effective arterial elastance
end‐diastolic pressure‐volume relationship
end‐systolic elastance
endothelin
fraction of inspired oxygen
interleukin‐6
inducible nitric oxide synthase
interventricular septum
left ventricle/left ventricular
monocrotaline
messenger ribonucleic acid
nitric oxide
nitric oxide synthase
pulmonary arterial hypertension
polymerase chain reaction
prostaglandin I2 or prostacyclin
pulmonary hypertension
maximum pressure
oxygen partial pressure
reverse transcription
right ventricle/right ventricular
tumor necrosis factor‐α
Pulmonary hypertension (PH) is defined on hemodynamic grounds as mean pulmonary arterial pressure >25 mmHg. It is a syndrome with different etiologies and represents the most serious chronic disease of pulmonary circulation. PH encompasses a group of disorders characterized by pulmonary vascular remodeling that leads to right ventricular (RV) hypertrophy and dysfunction. Less than a decade ago, treatment was essentially empirical and ineffective and the natural history of the disease was associated with high mortality. Despite recent advances in therapy, which have contributed to improvements in quality of life and survival of these patients, vasodilator therapy is insufficient and PH is still associated with a poor prognosis. In severe forms, PH progresses independently of treatment to RV dysfunction, which is the most important predictor of mortality. In refractory cases, lung or combined heart‐lung transplantation are the last therapeutic options. In addition to myocardial involvement, severe PH also has systemic effects, mainly due to inflammatory activation, which contributes to morbidity and mortality. New therapeutic approaches, acting at systemic and myocardial levels, are therefore required to prevent clinical deterioration.1 Additionally, the molecular mechanisms of pulmonary vasodilators remain to be clarified, in particular their molecular mechanisms of action and their functional consequences at myocardial and systemic levels. Furthermore, most pulmonary artery vasodilators also exert negative inotropic effects and lead to systemic vasodilation,2 which can be harmful, especially in patients with established RV dysfunction. Moreover, pulmonary artery vasodilation may exacerbate ventilation‐perfusion mismatch.3
Endothelin‐1 (ET‐1) antagonists are now widely used in the chronic treatment of most forms of pulmonary arterial hypertension (PAH) and off‐label in many other forms of PH. The dual ET‐1 receptor antagonist bosentan is effective in patients with PH,4 showing sustained long‐term effects.5 It improves oxygenation and functional status in adults with Eisenmenger syndrome and PH associated with congenital heart disease.6 Bosentan has been approved by the US Food and Drug Administration for the treatment of World Health Organization functional classes III and IV. It improves 6‐minute walk test results, delays clinical deterioration and improves survival in idiopathic PAH compared to historical cohorts.7 Despite the recognized clinical benefit of ET‐1 antagonism, its functional and molecular effects at the myocardial level are still not well characterized. Although some experimental8 and clinical9 reports suggest an improvement in myocardial performance in PH after chronic bosentan therapy, this hypothesis has not been tested.
In order to clarify its RV myocardial and systemic actions, we assessed the effect of chronic bosentan administration in an experimental model of PH induced by monocrotaline (MCT) by performing hemodynamic and morphological assessment, blood gas exams and molecular biology studies.
MethodsAnimal modelMale Wistar rats weighting 180–200 g (Charles River Laboratories, Barcelona, Spain) randomly received a subcutaneous injection of 60 mg/kg MCT or an equivalent volume of vehicle (Ctrl, n=8). Forty‐eight hours later, MCT‐injected animals were again randomized into one subgroup that was treated daily with 300 mg/kg bosentan (30 mg/ml in 5% gum arabic) by gavage (MCT‐BOS, n=14) and another that received a corresponding volume of vehicle (MCT, n=35). Animals were housed in groups of five per cage under controlled conditions, with a light‐dark cycle of 12:12 hours and a constant temperature of 22°C, and water and food ad libitum (experimental protocol summarized in Figure 1). Bosentan was kindly provided by Actelion Pharmaceuticals. Experimental procedures were performed in accordance with Portuguese law on animal welfare and the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH publication no. 85‐23, revised 1996).
Hemodynamic evaluation MCT: monocrotaline‐injected rats;
MCT: monocrotaline‐injected rats; In the fifth week after MCT injection, animals were anesthetized with 8% sevoflurane inhalation (Penlon Sigma Delta Anesthetic Vaporizer), orotracheally intubated and mechanically ventilated with 100% oxygen. Ventilatory frequency was programmed for 150/min and tidal volume adjusted to animal weight, following the manufacturer's instructions (TOPO Small Animal Ventilator, Kent Scientific Corporation). Anesthesia was maintained with sevoflurane at the minimum inspired fraction required to suppress the toe pinch reflex (2.5 to 3%). Continuous ECG (DII), pulse oximetry, and rectal temperature (maintained at 38°C) were monitored and blood volume was restored through the dorsal foot vein (Multi‐Phaser NE‐1000 syringe pump, New Era Pump Systems, Wantagh, NY), with warm Ringer's lactate infusion, at a rate of 32 ml/kg/h. Left thoracotomy and pericardiectomy were performed under a dissecting microscope (Wilde M651, Leica Microsystems, Cambridge, UK). Pressure‐volume catheters were inserted through the apex, along the long axes of the ventricles (SPR‐838 and PVR‐1045, Millar Instruments, Houston, TX, for the left ventricle (LV) and RV, respectively). A transit time flow probe was inserted in the ascending aorta to measure cardiac output (CO) (200–367, Triton Technology). Arterial blood was collected for gas analysis (Stat Profile pHOx®, Nova Biomedical) and ventilator parameters were adjusted to achieve normocapnia. After stabilization for 15 minutes, recordings were made with ventilation suspended at end‐expiration for the baseline condition and during transient inferior vena cava occlusion. Parallel conductance and field inhomogeneity (factor α) were determined by 50‐μl injections of hypertonic saline solution (NaCl 10%) and direct measurement of CO, respectively. Data were continuously acquired (MPVS 300, Millar Instruments), digitally recorded at 2000 Hz (ML880 PowerLab 16/30, ADInstruments) and analyzed offline (PVAN™ 3.5, Millar Instruments). On completion of the assessment, the rats were euthanized by exsanguination under anesthesia overdose. The volume signal was calibrated in a cuvette with standard wells using heparinized blood. A non‐heparinized 2‐ml blood sample was collected and stored at −20°C after centrifugation. The RV, LV and interventricular septum (IVS) were weighed, and samples collected from the free walls of both ventricles were immediately frozen in liquid nitrogen and stored at −80°C.
Gene expressionTotal messenger ribonucleic acid (mRNA) was extracted from the RV myocardium by the guanidinium thiocyanate and silica membrane method (Qiagen, no. 74124), according to the manufacturer's instructions. Concentration and purity were determined by spectrophotometry (Eppendorf, no. 6131000.012). Relative mRNA quantification was performed by reverse transcription (RT) and real‐time polymerase chain reaction (PCR). RT was performed in a thermocycler (Whatman Biometra, no. 050‐901) using 30 ng/μl random primers (Invitrogen, no. 48190‐011) and 30 U/reverse transcriptase (Invitrogen, no. 18064‐014), in a final volume of 20 μl. Complementary deoxyribonucleic acid obtained was then amplified by real‐time PCR (LightCycler II, Roche), using SYBR Green (Quantitect, no. 204143) as a marker, according to the manufacturer's instructions. Probes were designed in‐house with the aid of software and specific amplification of the target genes was confirmed in ethidium bromide gel (Table 1). For each gene, standard curves were created by serial dilution of a randomly selected tissue sample (R>0.99). In each experiment, a similar amount of mRNA from each sample was simultaneously amplified, and the standard curve analysis was determined from the correlation between the starting amount of total mRNA and the PCR threshold cycle (second derivative maximum method) of graded dilutions. Experiments were carried out in duplicate. β‐actin was used as an internal control of gene expression. Results are expressed in relative terms as arbitrary units (AU) compared with Ctrl.
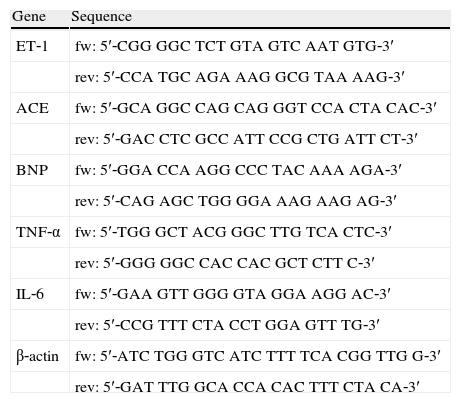
Specific probe sequences for forward (fw) and reverse (rev) amplification by polymerase chain reaction.
| Gene | Sequence |
| ET‐1 | fw: 5′‐CGG GGC TCT GTA GTC AAT GTG‐3′ |
| rev: 5′‐CCA TGC AGA AAG GCG TAA AAG‐3′ | |
| ACE | fw: 5′‐GCA GGC CAG CAG GGT CCA CTA CAC‐3′ |
| rev: 5′‐GAC CTC GCC ATT CCG CTG ATT CT‐3′ | |
| BNP | fw: 5′‐GGA CCA AGG CCC TAC AAA AGA‐3′ |
| rev: 5′‐CAG AGC TGG GGA AAG AAG AG‐3′ | |
| TNF‐α | fw: 5′‐TGG GCT ACG GGC TTG TCA CTC‐3′ |
| rev: 5′‐GGG GGC CAC CAC GCT CTT C‐3′ | |
| IL‐6 | fw: 5′‐GAA GTT GGG GTA GGA AGG AC‐3′ |
| rev: 5′‐CCG TTT CTA CCT GGA GTT TG‐3′ | |
| β‐actin | fw: 5′‐ATC TGG GTC ATC TTT TCA CGG TTG G‐3′ |
| rev: 5′‐GAT TTG GCA CCA CAC TTT CTA CA‐3′ |
ACE: angiotensin‐converting enzyme; BNP: B‐type natriuretic peptide; ET‐1: endothelin‐1; IL‐6: interleukin‐6; TNF‐α: tumor necrosis factor‐α.
The differential activity of cyclooxygenase (COX) and nitric oxide synthase (NOS) was quantified in RV myocardium homogenates (760151 and 760871, Cayman Chemical Company, respectively), with and without COX‐2 (DuP‐697; 70645, Cayman Chemical Company) and inducible NOS (iNOS) (aminoguanidine; 396494, Sigma‐Aldrich) inhibitors. Residual activity was assessed by heat inactivation. In the NOS activity quantification assay, preliminary ultrafiltration was carried out in order to concentrate proteins and remove tissue nitrates (Amicon Ultra 30K, Millipore).
Plasma mediatorsPlasma levels of ET‐1 (S‐1171, Peninsula Laboratories), interleukin‐6 (IL‐6) (DE4845, Demeditec Diagnostics GmbH) and tumor necrosis factor‐α (TNF‐α) (45‐TNFRTU‐E01, ALPCO Diagnostics) were quantified by quantitative enzyme immunoassay (UVM‐340 monochromator based reader, ASYS Hitech GmbH). After ultrafiltration (Amicon Ultra 30K, Millipore), stable nitric oxide (NO) and prostacyclin (PGI2) metabolites, nitrates and 6‐keto‐prostaglandin F1α (6‐keto‐PGF1α) were also quantified (Cat. no. 760871 and Cat. no. 515211, Cayman Chemical Company, respectively).
Statistical analysisOne‐way ANOVA or ANOVA on ranks tests were used for normal and non‐normal distributions, respectively, and the chi‐square test was used to compare survival. Results are expressed as mean ± standard error of mean. Statistical significance was assumed at a two‐tailed p value of <0.05.
ResultsAnimal modelMCT‐injected rats gradually developed weight loss and clinical signs of decompensated heart failure, particularly lethargy, tachypnea, jugular vein distention, pleural effusion, ascites, and signs of respiratory distress, such as intercostal inspiratory retraction. MCT‐injected rats chronically treated with bosentan showed less lethargy. None of the Ctrl rats died during follow‐up, while in MCT and MCT BOS mortality was 77% and 43%, respectively (p<0.001).
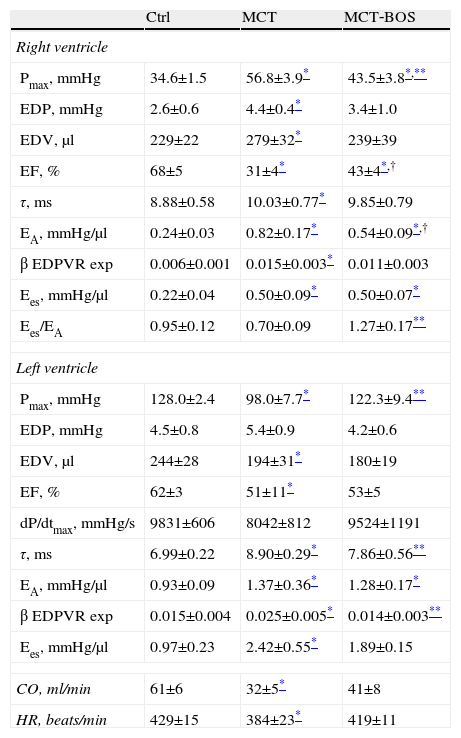
Hemodynamic evaluation, morphometry and arterial blood gas analysisThe hemodynamic parameters are summarized in Table 2 and representative pressure‐volume loops and end‐systolic and end‐diastolic pressure‐volume relationships are shown in Figure 2.
Right and left ventricular baseline and preload derivative variations obtained from inferior vena cava occlusions.
| Ctrl | MCT | MCT‐BOS | |
| Right ventricle | |||
| Pmax, mmHg | 34.6±1.5 | 56.8±3.9* | 43.5±3.8*,** |
| EDP, mmHg | 2.6±0.6 | 4.4±0.4* | 3.4±1.0 |
| EDV, μl | 229±22 | 279±32* | 239±39 |
| EF, % | 68±5 | 31±4* | 43±4*,† |
| τ, ms | 8.88±0.58 | 10.03±0.77* | 9.85±0.79 |
| EA, mmHg/μl | 0.24±0.03 | 0.82±0.17* | 0.54±0.09*,† |
| β EDPVR exp | 0.006±0.001 | 0.015±0.003* | 0.011±0.003 |
| Ees, mmHg/μl | 0.22±0.04 | 0.50±0.09* | 0.50±0.07* |
| Ees/EA | 0.95±0.12 | 0.70±0.09 | 1.27±0.17** |
| Left ventricle | |||
| Pmax, mmHg | 128.0±2.4 | 98.0±7.7* | 122.3±9.4** |
| EDP, mmHg | 4.5±0.8 | 5.4±0.9 | 4.2±0.6 |
| EDV, μl | 244±28 | 194±31* | 180±19 |
| EF, % | 62±3 | 51±11* | 53±5 |
| dP/dtmax, mmHg/s | 9831±606 | 8042±812 | 9524±1191 |
| τ, ms | 6.99±0.22 | 8.90±0.29* | 7.86±0.56** |
| EA, mmHg/μl | 0.93±0.09 | 1.37±0.36* | 1.28±0.17* |
| β EDPVR exp | 0.015±0.004 | 0.025±0.005* | 0.014±0.003** |
| Ees, mmHg/μl | 0.97±0.23 | 2.42±0.55* | 1.89±0.15 |
| CO, ml/min | 61±6 | 32±5* | 41±8 |
| HR, beats/min | 429±15 | 384±23* | 419±11 |
One‐way ANOVA; n=8 animals per group. CO: cardiac output; Ctrl: control rats; EA: effective arterial elastance; EA/Ees: ventriculo‐vascular coupling assessed by the ratio between end‐systolic elastance and arterial elastance; EDP: end‐diastolic pressure; EDV: end‐diastolic volume; Ees: end‐systolic elastance; EF: ejection fraction; Ees: slope of the relationship between end‐systolic pressure‐volume relationship and maximum or end‐systolic elastance; HR: heart rate; MCT: monocrotaline‐injected rats; MCT BOS: monocrotaline‐injected bosentan‐treated rats; Pmax: maximum or systolic pressure; β EDPVR exp: stiffness constant derived from the exponential end‐diastolic pressure‐volume relationship; τ: time constant of isovolumetric relaxation.
 MCT: monocrotaline‐injected rats;
MCT: monocrotaline‐injected rats; Representative pressure‐volume loops and respective end‐diastolic and end‐systolic pressure‐volume relationships, obtained by transient inferior vena cava occlusion. Ctrl: control group; MCT: monocrotaline‐injected rats; MCT BOS: monocrotaline‐injected bosentan‐treated rats. *p<0.05 vs. Ctrl by one‐way ANOVA; n=8 animals per group.
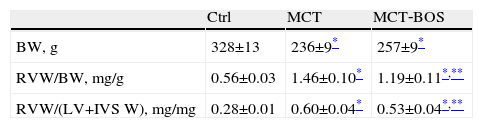
Compared with Ctrl, MCT‐injected groups developed PH, as assessed by higher RV systolic pressure, and their RV was subjected to increased afterload, as shown by increased effective pulmonary arterial elastance (EA) (Table 2). Following PH, both MCT‐injected groups had signs of RV failure, such as increased end‐diastolic volume, decreased ejection fraction, increased end‐diastolic pressure, prolonged time constant τ and an upward shift in the end‐diastolic pressure‐volume relationship (β EDPVR), reflecting greater stiffness. Chronic therapy with bosentan attenuated all the aforementioned changes, as manifested by the absence of difference between MCT‐BOS and Ctrl or significant attenuation compared with MCT. Of note, although both MCT‐injected groups showed higher contractility as assessed by load‐independent indices, end‐systolic elastance (Ees) given by the slope of the end‐systolic pressure‐volume relationship, and preload recruitable stroke work, MCT‐BOS presented significantly decreased EA compared with MCT. Therefore, the Ees/EA ratio, which reflects ventriculo‐vascular coupling, increased significantly in MCT‐BOS compared with MCT. Beyond this improvement, MCT‐BOS, unlike MCT, did not present decreases in CO or heart rate compared with Ctrl. Regarding RV morphometry and in accordance with greater afterload, we found a significant increase in RV to LV plus IVS weight ratio, indicating hypertrophy in both MCT‐injected groups, which was attenuated in MCT‐BOS (Table 3).
Morphology results obtained by necropsy.
| Ctrl | MCT | MCT‐BOS | |
| BW, g | 328±13 | 236±9* | 257±9* |
| RVW/BW, mg/g | 0.56±0.03 | 1.46±0.10* | 1.19±0.11*,** |
| RVW/(LV+IVS W), mg/mg | 0.28±0.01 | 0.60±0.04* | 0.53±0.04*,** |
n=8 per group. BW: body weight; Ctrl: control rats; LV+IVS W: left ventricle plus interventricular septum weight; MCT: monocrotaline‐injected rats; MCT BOS: monocrotaline‐injected bosentan‐treated rats; RVW: right ventricular weight.
As regards the LV, MCT presented decreased maximum developed pressure (Pmax) and concomitantly decreased end‐diastolic volumes and prolonged time constant of isovolumetric relaxation, τ, compared with Ctrl, suggesting impaired filling due to ventricular interaction. These changes were also attenuated in MCT‐BOS.
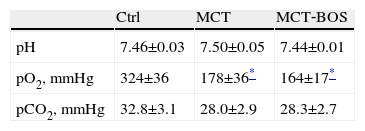
Both MCT and MCT‐BOS presented cachexia, with a significant decrease in body weight (Table 3). Regarding blood gas values (Table 4), both monocrotaline‐injected groups had a decrease in arterial oxygen partial pressure (pO2) compared with Ctrl, indicating respiratory distress. No significant differences secondary to treatment with bosentan were observed.
Arterial gas exam.
| Ctrl | MCT | MCT‐BOS | |
| pH | 7.46±0.03 | 7.50±0.05 | 7.44±0.01 |
| pO2, mmHg | 324±36 | 178±36* | 164±17* |
| pCO2, mmHg | 32.8±3.1 | 28.0±2.9 | 28.3±2.7 |
One‐way ANOVA; n=8 animals per group. Ctrl: control rats; MCT: monocrotaline‐injected rats; MCT BOS: monocrotaline‐injected bosentan‐treated rats; pO2: arterial oxygen partial pressure; pCO2: arterial carbon dioxide partial pressure.
Compared with Ctrl, MCT‐injected rats presented increased expression of B‐type natriuretic peptide (BNP), angiotensin‐converting enzyme (ACE), ET‐1 and TNF‐α, signaling overload and myocardial remodeling towards a fetal phenotype (Figure 3). These changes were attenuated in MCT‐BOS (Figure 3).
 MCT; one‐way ANOVA; n=8 animals per group.
MCT; one‐way ANOVA; n=8 animals per group. Right ventricular myocardium gene expression. Gene expression was normalized for β‐actin internal control gene expression, which did not differ between groups. Results are relative to an arbitrary unit set as the mean of Ctrl. *p<0.05 vs. Ctrl; **p<0.05 vs. MCT; one‐way ANOVA; n=8 animals per group. ACE: angiotensin‐converting enzyme; AU: arbitrary units; BNP: B‐type natriuretic peptide; Ctrl: control group; ET‐1: endothelin‐1; IL‐6: interleukin‐6; MCT: monocrotaline‐injected rats; MCT BOS: monocrotaline‐injected bosentan‐treated rats; mRNA: messenger ribonucleic acid.
MCT had increased plasma levels of ET‐1, IL‐6 and TNF‐α compared with Ctrl, which were attenuated in MCT‐BOS (Figure 4).
 MCT; one‐way ANOVA; n=8 animals per group. Ctrl: control group; ET‐1: endothelin‐1; IL‐6: interleukin‐6;
MCT; one‐way ANOVA; n=8 animals per group. Ctrl: control group; ET‐1: endothelin‐1; IL‐6: interleukin‐6; Plasma quantification of TNF‐α, IL‐6 and ET‐1. *p<0.05 vs. Ctrl; **p<0.05 vs. MCT; one‐way ANOVA; n=8 animals per group. Ctrl: control group; ET‐1: endothelin‐1; IL‐6: interleukin‐6; MCT: monocrotaline‐injected rats; MCT BOS: monocrotaline‐injected bosentan‐treated rats; TNF‐α: tumor necrosis factor‐α.
COX‐1 and ‐2 activity was increased in MCT RV myocardium compared with Ctrl, but was reduced after chronic bosentan treatment (Figure 5). Paradoxically, plasma levels of the stable PGI2 metabolite 6‐keto‐PGF1a were increased in both MCT and MCT‐BOS compared with Ctrl. In contrast, nitrate plasma levels were decreased only in MCT and were completely restored in MCT‐BOS, which, in fact, presented higher values than Ctrl. Plasma nitrate changes were paralleled by lower non‐inducible NOS activity, corresponding to endothelial and neuronal fractions, and by marked elevation of inducible NOS activity in MCT RV myocardium compared with Ctrl. These differences were significantly attenuated in MCT‐BOS (Figure 6).
 MCT; one‐way ANOVA; n=8 animals per group.
MCT; one‐way ANOVA; n=8 animals per group. COX‐1 and ‐2 activity (left and right upper panels, respectively) in RV and circulating levels (lower panel) of the stable metabolite 6‐keto‐PGF1α. *p<0.05 vs. Ctrl; **p<0.05 vs. MCT; one‐way ANOVA; n=8 animals per group. COX‐1: cyclooxygenase type 1; COX‐2: cyclooxygenase type 2; Ctrl: control rats; MCT: monocrotaline‐injected rats; MCT BOS: monocrotaline‐injected bosentan‐treated rats; PGF: 6‐keto‐prostaglandin F1α; RV: right ventricular myocardium.
 NOS activity (left and right upper panels, respectively) in RV and circulating levels (lower panel) of nitrates, which are stable metabolites of nitric oxide. *p<0.05 vs. Ctrl; **p<0.05 vs.
NOS activity (left and right upper panels, respectively) in RV and circulating levels (lower panel) of nitrates, which are stable metabolites of nitric oxide. *p<0.05 vs. Ctrl; **p<0.05 vs. Inducible and non‐inducible NOS activity (left and right upper panels, respectively) in RV and circulating levels (lower panel) of nitrates, which are stable metabolites of nitric oxide. *p<0.05 vs. Ctrl; **p<0.05 vs. MCT; one‐way ANOVA; n=8 animals per group. Ctrl: control rats; MCT: monocrotaline‐injected rats; MCT BOS: monocrotaline‐injected bosentan‐treated rats; NOS: nitric oxide synthase; RV: right ventricular myocardium.
This work demonstrates that chronic treatment with bosentan attenuates PH without exacerbating ventilation‐perfusion mismatch or causing systemic arterial hypotension. On the contrary, bosentan increases cardiac output and improves ventriculo‐vascular coupling. Additionally, there is an improvement in survival and an attenuation of RV myocardial and systemic inflammatory response.
MCT induces obliterative vasculitis, resulting in endothelial dysfunction, increased media thickness and loss of peripheral arterioles and arteries, leading to increased pulmonary vascular resistance, pressure overload and RV hypertrophy.10–14 This model has histopathological similarities with human primary PH progressing rapidly to RV failure. Following administration of MCT we observed PH, RV hypertrophy with dilation, diastolic dysfunction and myocardial molecular remodeling, inflammatory activation, cachexia and increased mortality. Chronic treatment with bosentan attenuated most of these changes. Bosentan, a dual ET‐1 antagonist, counteracts the important pathogenic role of ET‐1 in PH progression. In patients chronically treated with bosentan there is recovery of pulmonary vascular endothelial NOS activity and therefore higher nitrate plasma levels,15 whereas in MCT‐induced PH the opposite occurs.16 Corroborating these data, MCT‐BOS presented higher nitrate plasma levels. However, the pulmonary vasodilator effect of bosentan could worsen ventilation‐perfusion mismatch. Furthermore, since ET‐1 is a vasoconstrictor and a potent positive inotrope, its antagonism could be predicted to lead not only to systemic arterial hypotension, but also to ventriculo‐vascular uncoupling in the RV‐pulmonary circulation unit. Finally, functional antagonism of a mediator usually results in an increase in its plasma levels by feedback. Therefore we expected an increase in circulating ET‐1 levels. Interestingly, in MCT‐BOS none of these effects was found. The PO2/fraction of inspired oxygen (pO2/FiO2) ratio, usually taken as an oxygenation indicator during mechanical ventilation, was not compromised and therefore ventilation‐perfusion coupling was not impaired. There was an increase in cardiac output and an improvement in ventriculo‐vascular coupling, suggesting that chronic ET‐1 antagonism exerts a positive inotropic effect. Previous works suggested that chronic over‐activation of ET‐1 has negative inotropic effects and thus ET‐1 antagonism may have a positive inotropic effect in the failing myocardium.17 The molecular mechanisms that underlie this change in biological behavior are not well understood.18 Similar findings in ventriculo‐vascular coupling have been described in a model of PH induced by overflow.19 Similarly, LV systolic pressure increased in MCT‐BOS, indicating that chronic bosentan treatment does not cause systemic arterial hypotension. This may be due to improvement in cardiac output as well as attenuation of PH, which have a favorable effect on ventricular interaction, but may also be due to improvement in intrinsic LV myocardial function, as previously suggested.8,9 Finally, unlike as reported for acute ET‐1 antagonism by fast‐acting intravenous antagonists such as tezosentan,20 chronic bosentan treatment was not associated with increased ET‐1 plasma levels. This finding can be interpreted in light of the fact that in the setting of attenuated PH there is also a reduction of myocardial ET‐1 expression.12 Likewise, clinical improvement and reduction in RV afterload resulted in decreased expression of ACE and BNP.
RV myocardial inflammatory activation was studied, since it plays an important pathophysiological role in this experimental model. There was overexpression of TNF‐α in MCT RV myocardium and also an increase in TNF‐α and IL‐6 plasma levels in rats with PH. The MCT RV myocardium showed increased iNOS and COX‐2 activity. iNOS is responsible for production of large amounts of nitric oxide and mediates the negative inotropic effects of cytokines,21 while COX‐2, through the production of thromboxane A2,22 is partly responsible for the negative inotropic effects of TNF‐α in the failing myocardium.23 Chronic treatment with bosentan attenuated the inflammatory response. In fact, ET‐1 antagonism has direct anti‐inflammatory effects,24 which may partly explain the improvement in myocardial function of MCT‐BOS. Enhanced myocardial function and maintenance of cardiac output may also have contributed to improvement in tissue perfusion, attenuating inflammatory activity.25 The interaction between ET‐1 and physiological production of nitric oxide and prostanoids means they should be seen as complementary mechanisms. MCT RV myocardium showed a significant reduction in constitutive NOS activity, which produces nitric oxide in the myocardium at physiological levels,26 and increased COX‐1 activity, which is commonly activated by oxidative stress27 and ET‐1.28 Treatment with bosentan preserved constitutive NOS activity and antagonized excessive COX‐1 activity, which has been associated with protective effects in the failing myocardium.26,27
ConclusionThe present work clarifies the myocardial mechanisms of action of ET‐1 antagonists in an experimental model of PH. Unlike the physiological effects of ET‐1 in healthy myocardium, in overloaded, hypertrophied and failing RV myocardium of rats with PH, chronic ET‐1 antagonism has marked anti‐inflammatory effects that contribute to improvement in diastolic function and cardiac output and preservation of ventriculo‐vascular coupling. Pulmonary vasodilation did not worsen ventilation‐perfusion mismatch and, paradoxically, plasma ET‐1 levels were reduced. The clinical success of ET‐1 antagonists in the chronic treatment of PH, compared to other pulmonary vasodilators, may be due in part to complementary beneficial myocardial effects.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data and that all the patients included in the study received sufficient information and gave their written informed consent to participate in the study.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
Conflicts of interestThe authors have no conflicts of interest to declare.
This work was supported by the Portuguese Foundation for Science and Technology [Grants PEst‐OE/SAU/UI0051/2014 and EXCL/BIM‐MEC/0055/2012, partially funded by FEDER through COMPETE] through the Cardiovascular R&D Unit and by European Commission [Grant FP7‐Health‐2010; MEDIA‐261409].
